Abstract
There is compelling evidence to indicate an important role for increased local renin–angiotensin system activity in the pathogenesis of cardiac hypertrophy and heart failure. Resveratrol is a natural polyphenol that activates SIRT1, a novel cardioprotective and longevity factor having NAD+-dependent histone deacetylase activity. We tested the hypothesis whether resveratrol could prevent from angiotensin II (Ang II)-induced cardiovascular damage. Four-week-old double transgenic rats harboring human renin and human angiotensinogen genes (dTGR) were treated for 4 weeks either with SIRT1 activator resveratrol or SIRT1 inhibitor nicotinamide. Untreated dTGR and their normotensive Sprague–Dawley control rats (SD) received vehicle. Untreated dTGR developed severe hypertension as well as cardiac hypertrophy, and showed pronounced cardiovascular mortality compared with normotensive SD rats. Resveratrol slightly but significantly decreased blood pressure, ameliorated cardiac hypertrophy and prevented completely Ang II-induced mortality, whereas nicotinamide increased blood pressure without significantly influencing cardiac hypertrophy or survival. Resveratrol decreased cardiac ANP mRNA expression and induced cardiac mRNA expressions of mitochondrial biogenesis markers peroxisome proliferator-activated receptor-γ coactivator (PGC-1α), mitochondrial transcription factor (Tfam), nuclear respiratory factor 1 (NRF-1) and cytochrome c oxidase subunit 4 (cox4). Resveratrol dose-dependently increased SIRT1 activity in vitro. Our findings suggest that the beneficial effects of SIRT1 activator resveratrol on Ang II-induced cardiac remodeling are mediated by blood pressure-dependent pathways and are linked to increased mitochondrial biogenesis.
Introduction
The renin–angiotensin system (RAS) plays a pivotal role in the pathogenesis of hypertension, cardiac hypertrophy and heart failure. Besides the classical regulatory effects on blood pressure, sodium excretion, and aldosterone secretion, there was accumulating evidence indicating that RAS induces inflammatory response and oxidative stress by blood pressure-independent mechanisms (Citation1–3). It was shown very recently that angiotensin II (Ang II), the key effector of the RAS, induces mitochondrial dysfunction via protein kinase C-dependent pathway (Citation4) and cellular senescence (Citation5).
Sirtuin1 (SIRT1) is a mammalian nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase (HDAC) that regulates the acetylation level of several regulatory proteins related to energy homeostasis, DNA repair, cell survival and lifespan extension. A study using SIRT1-defective mice has shown that SIRT1 is essential for normal development (Citation6). SIRT1 regulates energy metabolism and response to caloric restriction in mice (Citation6). SIRT1 deacetylates p53 and represses its activity in order to protect various cells from DNA damage-induced apoptosis (Citation7,Citation8). By modulating the activity of endothelial nitric oxide synthase (eNOS), FoxO1 and p53, SIRT1 also promotes vasodilatory and regenerative functions of endothelial and smooth muscle cells (Citation9).
The most potent natural compound capable to activate SIRT1, and thus to mimic the beneficial effects of caloric restriction is resveratrol (3,5,4′-trihydroxystilbene) (Citation10,Citation11). Resveratrol is a constituent of red wine that exerts cardioprotective effects and has been shown to extend lifespan via mechanisms linked to increased cellular stress resistance and decreased apoptosis (Citation11,Citation12).
Resveratrol has been shown to improve endothelial function and attenuate vascular inflammation in aged animal models where mitochondrial biogenesis, measured as the expression of PGC-1α (peroxisome proliferator-activated receptor receptor [PPAR]-γ coactivator 1α), NRF-1 (nuclear respiratory factor 1) and Tfam (mitochondrial transcription factor), is impaired. Several lines of evidence support the notion that mitochondrial dysfunction plays an important role in cellular senescence, oxidative stress, and vascular inflammation (Citation13–15).
The aim of this study was to investigate whether resveratrol could prevent from Ang II-induced cardiovascular damage and influence mitochondrial biogenesis. We conducted our experiment in double transgenic rats harboring human renin and human angiotensinogen genes (dTGR), a novel transgenic model of Ang II-induced hypertension and cardiac hypertrophy (Citation16,Citation17).
Materials and Methods
Experimental animals, dietary and drug regimens and sample preparation
We used 66 4-week-old male double transgenic rats harboring human renin and angiotensinogen genes (dTGR) (dTGR Biotechnology and Animal Breeding Division, Füllingsdorf, Switzerland) and six aged-matched normotensive Sprague–Dawley (SD) controls (Charles River, Schutzfeld, Germany). Development and characteristics of dTGR have been described elsewhere (Citation16,Citation17). The protocols were approved by the Animal Experimentation Committee of the University of Helsinki, Finland, and the Provincial State Office of Southern Finland (approval number STU 1187 A), whose standards correspond to those of the American Physiological Society. The rats had free access to chow (SDS Special Diet Services, Witham, Essex, UK) and drinking water. dTGR and normotensive SD control rats were divided into four groups to receive the following drug regimens for 4 weeks: (i) dTGR controls (n=40); (ii) dTGR±resveratrol (trans-3,5,4′-trihydroxystilbene, Orchid Chemicals & Pharmaceuticals Ltd., India; 800 mg/kg by gavage) (n=13); (iii) dTGR±nicotinamide (NAM, 3-pyridinecarboxamide, Sigma-Aldrich, Germany; 400 mg/kg i.p.) (n=13); (iv) SD controls (n=6).
Systolic blood pressure was measured weekly by using tail cuff blood pressure analyzer (Apollo-2AB Blood Pressure Analyzer, Model 179-2AB, IITC Life Science, Woodland Hills, CA, USA). Urine samples were collected over 24-h period in metabolic cages at week 7 for albumin measurement. Urine volumes and water intakes were measured gravimetrically. Rats were anesthetized with CO2/O2 (AGA, Riihimäki, Finland) and decapitated. Blood samples were collected for biochemical measurements using ethylenediamine tetraacetic acid (EDTA) as an anticoagulant. The hearts were excised, washed with ice-cold saline, blotted dry and weighed. Tissue samples were snap-frozen in liquid nitrogen. Cardiac samples were stored at −80°C until assayed. Cardiac tissue samples for immunostaining were snap-frozen in isopentane (−35°C).
Cardiomyocyte cross-sectional area
Cardiac samples for histology were fixed with 10% neutral buffered formalin solution for 24 h, then dehydrated and embedded in paraffin; 4–5μm sections were cut and stained with hematoxylin eosin staining. Conventional light microscopy at ×400 magnification was used to determine the degree of cardiomyocyte hypertrophy. The cross-sectional area was evaluated in a blinded fashion and was analyzed using the ISIimaging software (Image Solutions Inc., Whippany, New Jersey, USA) (Citation8).
Coronary and myocardial damage
Myocardial samples were evaluated without the knowledge of the treatments, with conventional light microscopy (Citation18). The severity of observed lesions was graded with numerical values denoting to the degree of damage at the whole tissue level. The following system of severity grading was used: 0, no abnormalities detected; 1, minimal; 2, mild; 3, moderate; 4, marked; or 5, severe. In the cardiac samples, severity grading was performed on the coronary arteries and the myocardium
Biochemical determinations
Urinary albumin was measured by ELISA using rat albumin as a standard (Calltrend, Luckenwalde, Germany), and serum aldosterone according the instructions of the manufacturer (Coat-a-Count Aldosterone RIA kit; DPC Biermann, Bad Nauheim, Germany).
Myocardial gene expression analysis by quantitative real-time reverse transcriptase PCR assay [qRT-PCR]
Myocardial mRNA expressions of SIRT1, atrial natriuretic peptide (ANP) (Citation19), PGC-1α, NRF-1, Tfam, cytochrome c oxidase subunit 4 (cox4), Ink4a (Citation20), Arf (Citation20) and 18s (Citation21) were determined by quantitative real-time RT-PCR (LightCycler, Roche Diagnostics, Neuilly Sur Seine, France). The following primers were used: SIRT1 forward GCAGACGTGGTAATGT reverse ACACTCTCCCCAGTAG; PGC-1α forward GGTCCCCAGGCAGTAG, reverse CTCCATCATCCCGCAG; NRF-1 forward GCTTGCGTCGTCTGGAT, reverse GCACCGTGTCGCTCAT; Tfam forward AGACCTCGGTCAGCATATAACA, reverse GCGACGGATGAGATCACTT; cox4 forward TGGGAGTGTTGTGAAGAGTGA, reverse GCAGTGAAGCCGATGAAGAAC; 18S forward CATCCAAGGAAGGCAGCAG, reverse TTTTCGTCACTACCTCCCCG.
The quantities of ANP, SIRT1, PGC-1α, NRF-1, Tfam, cox4, Ink4a, Arf and 18s PCR products were quantified with an external standard curve amplified from purified PCR product.
Myocardial SIRT1, p53, acetylated p53 and cardiomyocyte SIRT1 expressions by Western blot
Myocardial samples from the left ventricle and cardiomyocyte cells were electrophoretically separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (from 30 to 40 μg total protein per lane). Proteins were transferred to a polyvinyldifluoride membrane (Immobilon-FL; Millipore, Bedford, Massachusetts, USA) and blocked in 5% non-fat milk–Tris-buffered saline–0.01% Tween–20 buffer. The membranes were probed with the following antibodies; anti-Sir2alpha, 1/1000 (Upstate), antip53, 1/1000 (Chemicon, Temecula, California, USA), anti acetyl-p53, 1/1000 (K373/K382) (Upstate). Tubulin was used as the loading control (Antialpha tubulin; Abcam). Horseradish peroxidase–conjugated antirabbit secondary antibody (Chemicon) was subjected to enhanced chemiluminescence solution (ECLplus; Amersham Biosciences, Buckinghamshire, UK). We quantified the relative protein expression in separate samples from the membranes with a fluorescent image analyzer (Fujifilm Europe GmbH, Dusseldorf, Germany).
Cardiomyocyte cells studies
Neonatal rat cardiomyocytes were isolated from hearts of the embryonic Wistar rats (E17). Rats were decapitated and hearts were quickly excised. Hearts were minced and enzymatically digested in a buffer containing 120 mM NaCl, 1 mM NaH2PO4, 20 mM HEPES, 5.5 mM glucose, 5.4 mM KCl, 0.8 mM MgSO4, 1 mg/ml collagenase IV (Worthington Biochemical Corp, Lakewood, NJ) and 2.5% trypsin (Sigma-Aldrich. St. Louis, MO). Tissue was digested by incubating in a 37°C water bath for 20 min and supernatant with cells was collected, centrifuged and resuspended in fetal bovine serum (FBS). This cycle was repeated five times until all tissue was digested. After digestion, supernatant from digestion cycles was combined, centrifuged and resuspended in growth medium containing Dulbecco’s modified Eagle’s medium (DMEM), 5% FBS and 10% horse serum (all from Gibco). To remove non-myocyte cells, culture was plated on a cell culture dish for 90 min in growth medium and non-adherent cells (mostly myocytes) were collected and plated on 0.2% gelatin-coated 12-well plates for experiments. After 24 h, cells were washed and growth medium was replaced with a serum-free medium and incubated for another 24 h before treatments. The cell isolation method is same as described by Chlopčíková et al. (Citation22) with the exception of adding 1 mg/ml collagenase IV (Worthington) to digestion buffer. Cells were treated with Ang II (100 nM, Sigma-Aldrich) alone and Ang II with angiotensin II type 1 receptor (AT1R) antagonist losartan (10 μM, Sigma-Aldrich) or with AT2R antagonist PD123319 (10μM, Sigma-Aldrich) for 48 h. After incubation, cells were collected with Laemmli sample buffer (Biorad, Hercules, CA) for analyzing SIRT1 protein level in cells.
Measurement of SIRT1 activity
SIRT1 Fluorimetric Drug Discovery Kit (AK-555, Enzo Life Sciences Int., PA, USA) was used to measure SIRT1 activity. Human recombinant SIRT1 was incubated together with 25 μM Fluor de Lys-Sirt1, deacetylase substrate and 25 μM NAD+ (37°C), in the absence (Control) or presence of resveratrol (2, 10, 20, 40 and 100 μM) to produce deacetylated product. In the second step, treatment with Developer II/2 mM nicotinamide produced a fluorophore, which was detected on a fluorometric plate reader (Victor2 Multilabel Counter, PerkinElmer, Ex. 355 nm, Em. 460 nm).
Statistical analysis
Data are presented as the mean±SEM. Statistically significant differences in mean values were tested by analysis of variance (ANOVA) and Newman–Keul’s post hoc test or using an unpaired, two-tailed Student’s t-test when comparing only two groups. The differences were considered significant when p<0.05. The Kaplan–Meier test was used for survival analysis.
Results
Cardiovascular mortality
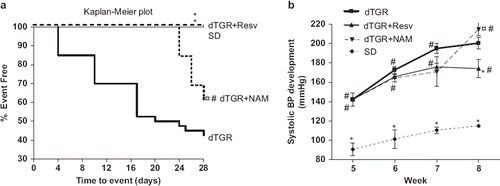
Only 43% of untreated dTGR (17/40) survived the 4-week follow-up period (), whereas none of the dTGR treated with resveratrol or SD controls died. NAM treatment slightly prevented cardiovascular mortality in dTGR (survival rate 62%, 8/13).
Figure 1. Survival curves of dTGR, dTGR treated with resveratrol, dTGR treated with nicotinamide and SD rats (a). The log rank test was used to compare the Kaplan–Meier survival curves with each other. *p<0.05 compared with dTGR; ¤p<0.05 compared with dTGR+Resveratrol; #p<0.05 compared with SD. Graph showing the effects of resveratrol and nicotinamide on systolic blood pressure development (b). Resveratrol significantly decreased systolic blood pressure compared with dTGR. dTGR denotes untreated double transgenic rats harboring human renin and angiotensin genes; dTGR+Resv denotes double transgenic rats harboring human renin and angiotensin genes treated with resveratrol 800 mg/kg; dTGR+NAM denotes double transgenic rats harboring human renin and angiotensin genes treated with nicotinamide 400 mg/kg; SD denotes normotensive Sprague–Dawley rats. Means±SEM are given, n=6–17 in each group. *p<0.05 compared with dTGR; ¤p<0.05 compared with dTGR+Resveratrol; #p<0.05 compared with SD.
Blood pressure, cardiac hypertrophy and cardiac morphology
Untreated dTGR developed pronounced hypertension () with cardiac hypertrophy assessed as heart weight-to-body weight ratio (). In the dTGR heart samples, lesions of the cardiac arteries varied from minimally thickened media and adventitia to moderate hyperplasia of intimal/medial layers and fibrosis of the adventitia with inflammatory cell infiltration. Lesions in the myocardial muscle were seen as accumulations of excess connective tissue and foci of inflammatory cells with elevated coronary damage () and myocardial damage scores ().
Figure 2. Bar graphs showing the effects of resveratrol and nicotinamide treatments on cardiac hypertrophy measured as heart weight-to-body weight ratio (a), cardiomyocyte cross-sectional area (b), coronary damage score (c), and myocardial damage score (d). For abbreviations, see Means±SEM are given, n=6–17 in each group. *p<0.05 compared with dTGR; ¤p<0.05 compared with dTGR+Resveratrol; #p<0.05 compared with SD.
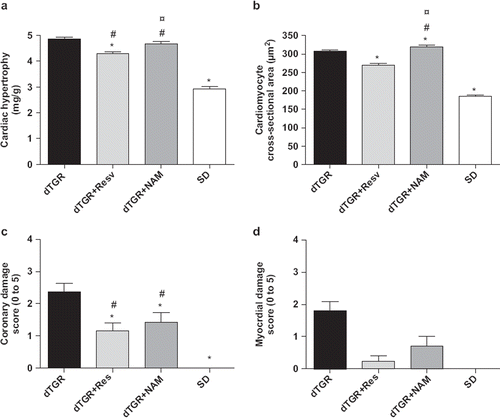
Resveratrol treatment produced a significant decrease in blood pressure by approximately 35 mmHg (), ameliorated cardiac hypertrophy measured as heart weight-to-body weight by 12% () and as cardiomyocyte cross-sectional area by 13% (). Resveratrol also partially prevented Ang II-induced coronary and myocardial damage ( and ). In contrast, NAM increased blood pressure in dTGR (), and induced an 8% increase in cardiomyocyte cross-sectional area () without significantly affecting heart weight-to-body weight ratio (). Surprisingly, NAM produced a slight but significant improvement in coronary and myocardial morphology compared with untreated dTGR ( and ). In SD controls, systolic blood pressure was 115±1 mmHg without any signs of cardiac hypertrophy or cardiac damage.
Cardiac ANP mRNA expression
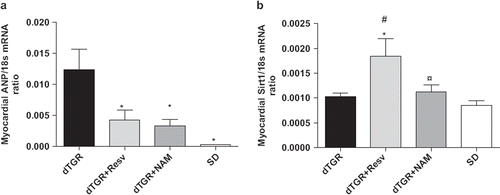
Cardiac ANP mRNA expression in untreated dTGR was increased to 63-fold as compared with SD controls. Resveratrol decreased cardiac ANP mRNA expression by 66%; however, the difference did not quite reach statistical significance. Surprisingly, also NAM produced a non-significant decreased in cardiac ANP mRNA expression ().
Figure 3. Bar graphs showing the effects of the 8-week resveratrol and nicotinamide treatments on myocardial ANP mRNA expression (a), and SIRT1 mRNA expression (b). For abbreviations, see Means±SEM are given, n=6–17 in each group. *p<0.05 compared with dTGR; ¤p<0.05 compared with dTGR+Resveratrol; #p<0.05 compared with SD.
Myocardial PGC-1α, NRF-1, Tfam and cox4 mRNA expressions
There were no differences between untreated dTGR and SD in the cardiac mRNA expressions of mitochondrial biogenesis markers PGC-1α, NRF-1, Tfam or cox4 (). Resveratrol produced a significant increase in cardiac PGC-1α, Tfam and cox4 mRNA levels. In contrast, NAM did not influence the mitochondrial biogenesis markers in the heart.
Figure 4. Bar graphs showing the effects of the 8-week resveratol and nicotinamide treatment on myocardial PGC–1α (a), NRF–1 (b), Tfam (c), and COX 4 (d); in panel (e) Ink4a and in panel (f) Arf mRNA expression between dTGR and SD rats. For abbreviations, see Means±SEM are given, n=6–17 in each group. *p<0.05 compared with dTGR; ¤p<0.05 compared with dTGR+resv; #p<0.05 compared with SD rats.
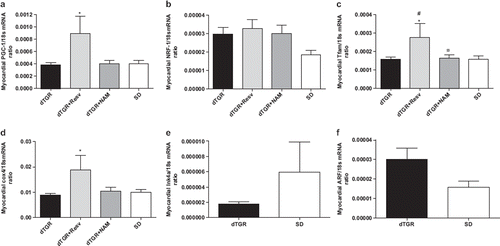
Myocardial Ink4a and Arf mRNA expressions
There were no statistically significant differences in the cellular senescence markers Ink4a and Arf between untreated dTGR and SD controls, although Arf mRNA expression was 48% greater in untreated dTGR ( and ).
Cardiac SIRT1 expression, P53 and acetylated-P53
Cardiac SIRT1 protein expression was higher in untreated dTGR compared with SD controls (), whereas there was no difference in cardiac SIRT1 mRNA expression indicating changes in post-transcriptional regulation (). Resveratrol decreased SIRT1 protein expression by 42% () but increased cardiac SIRT1 mRNA expression (). NAM decreased cardiac SIRT1 protein expression to a same extent as resveratrol () but did not influence cardiac SIRT1 mRNA expression ().
Figure 5. Bar graphs showing the effects of 8-week resveratrol and nicotinamide treatments on Sirt1 protein expression (a), myocardial Sirt1 protein expression in Dahl HS and Dahl SS rats (b), and Sirt1 protein expression in neonatal cardiomyocytes (c). dTGR denotes unmedicated double transgenic rats harboring human renin and angiotensin genes; Dahl HS denotes Dahl salt-sensitive rats on high salt diet (NaCl 0.8% w/w); Dahl LS denotes Dahl salt-sensitive rats on low salt diet (0.3% w/w). Means±SEM are given, n=6–17 in each group. *p<0.05 compared with dTGR.
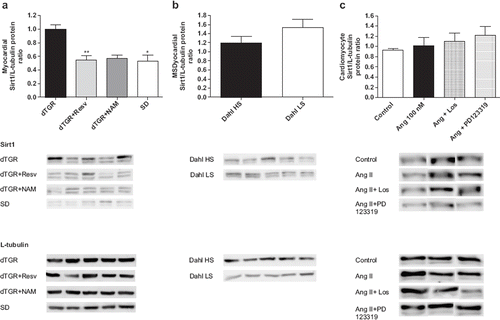
As a supplementary experiment, we examined cardiac SIRT1 expression from Dahl/Rapp salt-sensitive rats on high-salt diet (NaCl 8% w/w), a hypertensive animal model with low plasma renin activity and tissue RAS activity, as well as from their normotensive controls Dahl salt-sensitive rats on low-salt diet (NaCl 0.3% w/w). Development and characteristics of this inbred strain of Dahl salt-sensitive rat has been described in detail previously (Citation23,Citation24). There were no difference in cardiac SIRT1 expression between Dahl/Rapp rats receiving high-salt diet and low-salt diet (), although systolic blood pressure was markedly increased in Dahl/Rapp rats on high salt diet (systolic blood pressure 245±12 mmHg vs 164±11 mmHg, p<0.001).
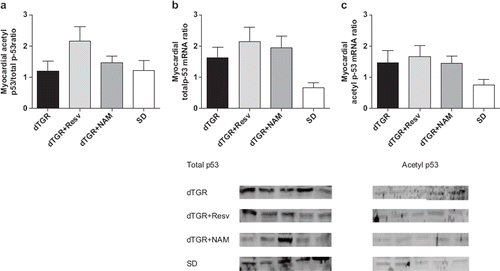
There were no differences in protein expression of cardiac P53, acetylated-P53 or acetylated-P53-to-P53 ratio ().
Figure 6. Bar graphs showing the effects of 8-week resveratrol and nicotinamide treatments on myocardial acetyl p53/total p53 protein expression (a), the effects of 8-week resveratrol and nicotinamide treatments on myocardial p53 total protein expression (b), and the effects of 8-week resveratrol and nicotinamide treatments on myocardial p53 acetyl protein expression (c). Means±SEM are given, n=6–17 in each group.
SIRT1 expression in rat neonatal cardiomyocytes
The effect of Ang II on SIRT1 protein level was studied using rat neonatal cardiomyocytes. There was no significant difference in SIRT1 protein level between Ang II and vehicle treated cells (). Blocking AT1R by losartan or AT2R by PD123319 did not influence SIRT1 protein level in rat neonatal cardiomyocytes ().
Effect of resveratrol and NAM on SIRT1 activity
Resveratrol increased Sirt1 activity by 17-fold in a dose-dependent manner. Nicotinamide (100 μM) decreased SIRT1 activity by more than 70% (data not shown).
Albuminuria and serum aldosterone level
Albuminuria was increased by 40-fold () and serum aldosterone level by sevenfold in untreated dTGR compared with SD controls (). Resveratrol decreased albuminuria by 25%; however, the difference did not reach statistical significance. Resveratrol significantly decreased serum aldosterone level by 50% compared with untreated dTGR. NAM did not influence albuminuria but it tended to increase serum aldosterone level.
Table I. Effects of resveratrol and nicotinamide on albuminuria and serum aldosterone level in double transgenic rats harboring human renin and angiotensinogen genes.
Discussion
There is now compelling evidence to indicate that Ang II induces vascular inflammation via blood pressure-independent mechanisms that are linked to increased oxidative stress and mitochondrial dysfunction. The important finding of the present study was that resveratrol, a mitochondrial biogenesis enhancer with anti-inflammatory and anti-oxidative properties (Citation25) completely prevented cardiovascular mortality in dTGR, a hypertensive animal model with increased local RAS activity. Resveratrol lowered blood pressure, ameliorated cardiac hypertrophy and decreased cardiac ANP mRNA expression, indicating reduced pressure/volume overload. Resveratrol also effectively induced mitochondrial biogenesis. In vitro studies using human recombinant SIRT1 protein revealed that resveratrol dose-dependently increased SIRT1 activity.
In line with the previous findings (Citation26,Citation27), cardiovascular mortality in dTGR is high reaching approximately 50–60% at the age of 8 weeks. We and others have shown previously that dTGR develop pronounced cardiac hypertrophy and heart failure with preserved systolic function, renal damage and vascular inflammation related to increased oxidative stress (Citation28). The detrimental effects of Ang II are mediated, at least in part, via blood pressure-independent mechanisms linked to induction of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase (Citation29). We here demonstrated for the first time, that resveratrol, a natural compound derived from the grapes, completely prevented Ang II-induced cardiac overmortality in dTGR. Resveratrol also decreased blood pressure and ameliorated cardiac hypertrophy assessed as heart weight-to-body weight ratio and cardiomyocyte cross-sectional area, and partially prevented from Ang II-induced coronary and myocardial damage. These findings indicate that the beneficial effects of resveratrol are likely to be mediated by blood-pressure dependent pathways. The markedly decreased cardiac ANP mRNA expression in resveratrol-treated dTGR strongly supports the notion on improved cardiac hemodynamics and decreased pressure/volume overload imposed to the heart. Interestingly we also noticed a 25% decrease in albuminuria in resveratrol-treated dTGR suggesting that resveratrol may also exert renoprotective properties.
SIRT1 is a member of sirtuin family of highly conserved NAD+-dependent enzymes regulating life span in lower organisms (Citation30). SIRT1 increases cellular stress resistance, genomic stability, and regulates cellular senescence and energy metabolism via deacetylation of the target proteins such as P53, FoxO transcription factors and PGC-1α. It has been claimed that the beneficial cellular effects of caloric restriction are largely mediated by induction of SIRT1 (Citation8,Citation31). Recently, Alcendor et al. (Citation32) demonstrated using a tissue-specific transgenic approach that moderate cardiac SIRT1 overexpression (2.5–7.5-fold increases) exerted cardioprotective effects that were linked to increased cellular stress resistance and, anti-apoptosis and protection against cellular senescence. In contrast, higher cardiac SIRT1 overexpression (more than 12.5-fold) was associated with detrimental effects such as development of heart failure, cardiac hypertrophy, apoptosis and increased oxidative stress. In the present study, cardiac SIRT1 expression was moderately increased in untreated dTGR compared with SD controls, whereas no difference was found in the cardiac SIRT1 mRNA expression. This finding suggests a change in the post-transcriptional regulation of SIRT1 in dTGR heart. To investigate further the regulation of cardiac SIRT1, we also examined the expression of SIRT1 from hypertensive Dahl/Rapp rats on high-salt diet, a widely used animal model of low-renin, low-Ang II hypertension. Although blood pressure and cardiac hypertrophy in Dahl/Rapp rats on high salt diet was at least as great as in untreated dTGR, we were unable to detect any differences in cardiac SIRT1 expression between hypertensive Dahl/Rapp rats on high salt diet and normotensive Dahl/Rapp rats on low-salt diet. We also conducted cell culture experiments using rat neonatal cardiomyocytes and were able to demonstrate that Ang II did not induce SIRT1 protein. Taken together, our findings suggest that neither hypertension nor Ang II alone is sufficient to induce cardiac SIRT1 expression, whereas a combination of pressure-induced overload together with high circulating/tissue Ang II levels induces cardiac SIRT1 protein expression. SIRT1 can thus be considered a novel cardioprotective factor participating in the regulation of Ang II-induced cardiac remodeling.
Accumulating evidence suggests that resveratrol acts as SIRT1 mimetic and induces changes in cellular metabolism similar to caloric restriction (Citation33). We therefore investigated the effects of resveratrol treatment on cardiac SIRT1 expression, cellular senescence and P53 acetylation. Unexpectedly, we were unable to detect difference in senescence markers Ink4a and ARF between dTGR and SD rats, neither any influence of oral resveratrol treatment on acetylation level of P53. In fact, cardiac SIRT1 expression was even lower in the resveratrol-treated dTGR compared with untreated controls. These findings were in contrast with data from our in vitro study using human recombinant SIRT1 protein, in which we demonstrated a dose-dependent increase in SIRT1 activity by resveratrol. However, it should be underlined that recent studies have raised concerns on the reliability of the Fluor-de-Lys assay for measurement of SIRT1 activity because of chemical interaction of the non-physiological fluorescent moiety of the peptide substrate with resveratrol (Citation34–36). Further studies are thus warranted to examine the effects of resveratrol on SIRT1 activity by employing biochemical assays utilizing native substrates lacking a fluoroprobe such as p53 or acetyl-CoA synthetase1. Taken together, our findings suggest that the beneficial cardiovascular effects of resveratrol in dTGR were mediated, at least in part, by SIRT1-independent pathways.
To investigate further the role of SIRT1 in the pathogenesis of Ang II-induced cardiac remodeling, we also examined the cardiovascular effects of the SIRT1 inhibitor NAM in dTGR. NAM produced a modest increase in blood pressure, which was associated with increased cardiomyocyte cross-sectional area. NAM slightly decreased cardiac SIRT1 expression. Surprisingly, NAM was capable of partially preventing Ang II-induced cardiovascular mortality and protected also against Ang II-induced cardiac damage. The cellular mechanisms of NAM-induced cardioprotection remained unclear; however, Stevens et al. reported recently, that besides being a SIRT1 inhibitor, NAM acts also as a weak PARP inhibitor, antioxidant and calcium modulator capable of improving energy status and inhibiting cell death in ischemic tissues (Citation37). Interestingly, PARP-1 has been shown to contribute to Ang II-induced cardiac fibrosis (Citation38), whereas PARP-1 deficient mice are protected from Ang II-induced cardiac hypertrophy (Citation39). Rajamohan et al. demonstrated very recently that SIRT1-dependent deacetylation inhibits PARP1 activity and protects cells from PARP1-mediated cell death (Citation40). These findings suggest a complex interplay between NAM, SIRT1 and PARP1-mediated cell death.
Ang II has been shown to induce mitochondrial oxidative damage and mitochondrial dysfunction via a protein kinase C-dependent pathway that activates NADPH oxidase and leads to formation of peroxynitrite (Citation4). We showed very recently mitochondrial dysfunction, distinct cardiac substrate utilization, and altered metabolomic profile in dTGR (Citation41). Resveratrol improves mitochondrial function and protects against metabolic diseases via PGC-1-dependent pathway (Citation42). We therefore examined the influence of resveratrol treatment on cardiac expression of mitochondrial biogenesis markers PGC-1α, Tfam, cox4 and NRF-1. In line with the previous findings (Citation24), we here demonstrated that resveratrol induced mitochondrial biogenesis in dTGR. Our findings thus support the notion that, the beneficial cardiovascular effects of resveratrol in dTGR are mediated partially via enhanced mitochondrial biogenesis and thus improved cardiac energy metabolism. In contrast, we were unable to detect any differences in mitochondrial biogenesis markers between dTGR and SD rats.
Ang II induces secretion of aldosterone from adrenal glands via AT1 receptor stimulation. In the present study, we found that resveratrol treatment was associated with markedly decreased serum aldosterone level in dTGR. Previous studies by Fiebeler et al. (Citation43) have demonstrated that aldosterone antagonists such as spironolactone effectively prevent the development of Ang II-induced cardiac and renal damage, as well as Ang II-induced vascular inflammation and oxidative stress, in dTGR. Miyazaki et al. showed recently using vascular smooth muscle cells that resveratrol down-regulates AT1 receptor expression (Citation44). It is also known that SIRT1 inhibitor NAM upregulates the expression of AT1R mRNA and reverses resveratrol-induced AT1R suppression (Citation44). It is therefore tempting to speculate that the beneficial effects of resveratrol found in the present study, could have been mediated, at least in part, via down-regulation of the AT1 receptors and decreased aldosterone secretion. Further studies are warranted to examine the interaction of resveratrol, aldosterone, and AT1 receptor expression.
In conclusion, data from the present study using a novel hypertensive transgenic rat line with increased RAS activity suggest that the beneficial effects of oral resveratrol treatment on Ang II-induced cardiovascular mortality and cardiac remodeling are mediated mainly by blood pressure-dependent pathways linked to increased mitochondrial biogenesis.
Acknowledgements
This study was supported by grants from the Academy of Finland, Päivikki and Sakari Sohlberg Foundation, Foundation of Cardiovascular Research and the Sigrid Jusélius Foundation. We are grateful to Mrs Anneli von Behr, Ms Nada Bechara-Hirvonen, and Ms Sari Laakkonen for expert technical assistance.
Declaration of interests: The authors state no conflict of interest.
References
- Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Med Sci Monit. 2005;11:RA194–RA205.
- Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258.
- Verlohren S, Muller DN, Luft FC, Dechend R. Immunology in hypertension, preeclampsia, and target-organ damage. Hypertension. 2009;54:439–443.
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496.
- Westhoff JH, Hilgers KF, Steinbach MP, Hartner A, Klanke B, Amann K, Melk A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension. 2008;52:123–129.
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, . SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3:e1759.
- Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784.
- Vahtola E, Louhelainen M, Merasto S, Martonen E, Penttinen S, Aahos I, . Forkhead class O transcription factor 3a activation and Sirtuin1 overexpression in the hypertrophied myocardium of the diabetic Goto-Kakizaki rat. J Hypertens. 2008;26:334–344.
- Borradaile NM, Pickering JG. NAD (+), sirtuins, and cardiovascular disease. Curr Pharm Des. 2009;15:110–117.
- Oliva J, French BA, Li J, Bardag-Gorce F, Fu P, French SW. Sirt1 is involved in energy metabolism: The role of chronic ethanol feeding and resveratrol. Exp Mol Pathol. 2008;85: 155–159.
- Ghosh HS. The anti-aging, metabolism potential of SIRT1. Curr Opin Investig Drugs. 2008;9:1095–1102.
- Alcaín FJ, Villalba JM. Sirtuin activators. Expert Opin Ther Pat. 2009;19:403–414.
- Shin SM, Cho IJ, Kim SG. Resveratrol protects mitochondria against oxidative stress through AMPK-mediated GSK3 {beta} inhibition downstream of poly[ADP-ribose]polymerase-LKB1 pathway. Mol Pharmacol. 2009;76:884–895.
- Hossain MB, Ji P, Anish R, Jacobson RH, Takada S. Polymerase 1 Interacts with nuclear respiratory factor 1 [NRF-1] and plays a role in NRF-1 transcriptional Regulation. J Biol Chem. 2009;284:8621–8632.
- Viña J, Gomez-Cabrera MC, Borras C, Froio T, Sanchis-Gomar F, Martinez-Bello VE, . Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61:1369–1374.
- Bohlender J, Fukamizu A, Lippoldt A, Nomura T, Dietz R, Ménard J, . High human renin hypertension in transgenic rats. Hypertension. 1997;29:428–434.
- Ganten D, Wagner J, Zeh K, Bader M, Michel JB, Paul M, . Species specificity of renin kinetics in transgenic rats harboring the human renin and angiotensinogen genes. Proc Natl Acad Sci USA. 1992;89:7806–7810.
- Louhelainen M, Vahtola E, Kaheinen P, Leskinen H, Merasto S, Kytö V, . Effects of levosimendan on cardiac remodeling and cardiomyocyte apoptosis in hypertensive Dahl/Rapp rats. Br J Pharmacol. 2007;150:851–861.
- Heyen JR, Blasi ER, Nikula K, Rocha R, Daust HA, Frierdich G, . Structural, functional, and molecular characterization of the SHHF model of heart failure. Am J Physiol Heart Circ Physiol. 2002;283:H1775–H1784.
- Bastide K, Guilly MN, Bernaudin JF, Joubert C, Lectard B, Levalois C, . Molecular analysis of the Ink4a/Rb1-Arf/Tp53 pathways in radon-induced rat lung tumors. Lung Cancer. 2009;63:348–353.
- Wellner M, Dechend R, Park JK, Shagdarsuren E, Al-Saadi N, Kirsch T, . Cardiac gene expression profile in rats with terminal heart failure and cachexia. Physiol Genomics. 2005;20:256–267.
- Chlopčíková S, Psotová J, Miketová P. Neonatal rat cardiomyocytes – A model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2001;145:49–55.
- Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1985;7:340–349.
- Walder RY, Morgan DA, Haynes WG, Sigmund RD, McClain AM, Stokes JB, . Genetic characterization of the “new” Harlan Sprague–Dawley Dahl salt-sensitive rats. Hypertension. 1996;27:546–551.
- Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, . Resveratrol induces mitochondrial biogenesis in endothelial cells, Am J Physiol Heart Circ Physiol. 2009;297:H13–H20.
- Müller DN, Mervaala EM, Dechend R, Fiebeler A, Park JK, Schmidt F, . Angiotensin II (AT (1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am J Pathol. 2000;157:111–122.
- Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, . Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693.
- Helkamaa T, Finckenberg P, Louhelainen M, Merasto S, Rauhala P, Lapatto R, . Entacapone protects from angiotensin II-induced inflammation and renal injury. J Hypertens. 2003;21:2353–2363.
- Yamamoto E, Kataoka K, Yamashita T, Tokutomi Y, Dong YF, Matsuba S, . Role of xanthine oxidoreductase in the reversal of diastolic heart failure by candesartan in the salt-sensitive hypertensive rat. Hypertension 2007;50:657–662.
- Finkel T, Deng CX, Mostoslavsky. Recent progress in the biology and physiology of sirtuins. Nature 2009;460: 587–591.
- Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, . CALERIE Pennington Team. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76.
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, . Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521.
- López-Lluch G, Irusta PM, Navas P, de Cabo R, Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008;43:813–819.
- Borra MT, Smith BC, Denu JM. Mechanisms of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280: 17187–17195.
- Kaeberlein M, McDonaugh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, . Substrate-specific activation of sirtuins by resveratrol. J Biol Chem 2005;280:17038–17045.
- Haigis MC, Sinclair DA. Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol. 2010;5: 253–295.
- Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, . Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther. 2007;320:458–464.
- J Huang D, Wang Y, Yang C, Liao Y, Huang K. Angiotensin II promotes poly[ADP-ribosyl]ation of c-Jun/c-Fos in cardiac fibroblasts. Mol Cell Cardiol. 2009;46:25–32.
- Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly (ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291:H1545–H1553.
- Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, . SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly (ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129.
- Mervaala E, Biala A, Merasto S, Lempiäinen J, Mattila I, Martonen E, . Metabolomics in angiotensin II-induced cardiac hypertrophy. Hypertension. 2010;55:508–515.
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, . Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006;127:1109–1122.
- Fiebeler A, Schmidt F, Müller DN, Park JK, Dechend R, Bieringer M, . Mineralocorticoid receptor affects AP-1 and nuclear factor-kappab activation in angiotensin II-induced cardiac injury. Hypertension 2001;37:787–793.
- Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008;28:1263–1269.