Abstract
Innate immunity is constructed around genetically encoded receptors that survey the intracellular and extracellular environments for signs of invading microorganisms. These receptors recognise the invader and through complex intracellular networks of molecular signaling, they destroy the threat whilst instructing effective adaptive immune responses. Many of these receptors, like the Toll-like receptors in particular, are well-known for their ability to mediate downstream responses upon recognition of exogenous or endogenous ligands; however, the emerging family known as the C-type lectin-like receptors contains many members that have a huge impact on immune and homeostatic regulation. Of particular interest here are the C-type lectin-like receptors that make up the Dectin-1 cluster and their intracellular signaling motifs that mediate their functions. In this review, we aim to draw together current knowledge of ligands, motifs and signaling pathways, present downstream of Dectin-1 cluster receptors, and discuss how these dictate their role within biological systems.
Keywords :
INTRODUCTION
Cells of the innate immune system possess an array of germ-line encoded receptors that are essential for mounting an effective immune response to pathogens. Upon pathogen recognition, these receptors initiate and instruct innate and adaptive immune responses, ultimately leading to the containment and destruction of the invading organisms. Interestingly, many receptors have identified self-ligands and play important roles in the control of homeostasis [Citation1, 2]. These receptors are expressed by the host as transmembrane and cytoplasmic proteins and act to communicate stimuli of foreign origin to cytoplasmic signaling cascades. They are known as pattern recognition receptors (PRRs) and, as hypothesised by Charles Janeway, are characterised by their ability to bind evolutionary-conserved motifs present on pathogens [Citation3]. These PRRs have been divided into four families: the Toll-like receptors (TLRs), nucleotide-oligomerisation domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-1 (RIG-1)-like receptors (RLRs) and the C-type lectin-like receptors (CTLRs). Targets for these receptors include cell wall components such as carbohydrates, yet recognisable structures can include proteins, lipids and nucleic acids to name a few. Recognition of these conserved microbial motifs, also known as pathogen-associated molecular patterns (PAMP), allows the host to detect non-self molecules and trigger immune defences [Citation4]. Secreted PRRs also exist and are able to modulate, but not directly induce, anti-microbial inflammatory responses. However, in this review, we will only focus on the PRRs able to directly induce intracellular signaling events.
Receptor activation following pathogen recognition on cell types such as dendritic cells (DCs) induces intracellular signaling that can trigger several cellular responses, including phagocytosis of the receptor-bound pathogen and induction of various patterns of cytokine and chemokine expression, which in turn modulate innate and adaptive inflammatory responses. Phagocytosis is an intricate process involving receptor-mediated signaling cascades as well as actin cytoskeleton remodelling. Upon internalisation of the pathogen into a vacuole known as the phagosome, the phagosome matures into an increasingly anti-microbial compartment through increasing hydrolytic enzymes and toxic metabolite concentrations as well as reduction of internal pH. Other mechanisms involved include the respiratory burst pathways, where the activation of the multimeric enzyme NADPH oxidase along with its translocation into the phagosome membrane reduces O2 forming reactive oxygen species (ROS) such as H2O2 [Citation5]. Aside from its involvement in second messenger signaling, ROS is used as a microbicidal agent and is crucial for the degradation of the phagocytosed invader [Citation6]. Products of the ROS-induced degradation pathways can be processed and presented by DCs after phagocytosis, where detection of the non-self-ligands can enhance presentation of material from the phagosome. This process links the innate and adaptive immune responses, as antigen-presenting DCs expressing varying combinations of cytokines can migrate to the lymph nodes and activate naive T cells [Citation7]. Here, we will briefly discuss the four PRR families outlining their notable members, main functions, and signaling routes used, and then focus particularly on the Dectin-1 cluster of C-type lectins.
Toll-Like Receptors
The TLRs are considered to be the primary sensing receptors of the innate immune system and have been implicated in the defence against invading bacteria, fungi and viruses, amongst others. TLRs are the most extensively studied family of PRRs and, to date, 13 members have been identified. TLR1–9 are present in both humans and mice, whereas TLR10 and TLR11 are expressed in humans and mice, respectively, both TLR12 and TLR13 functions and expression profiles remain undefined. TLRs are type I transmembrane receptors containing a horseshoe-shaped ligand-binding domain (LBD) and a signaling tail connected by a transmembrane region. The signaling domain has structural homology to interleukin-1 receptor (IL-1R) and is subsequently called the Toll/IL-1 receptor (TIR) domain [Citation8, 9]. Functions of TLRs appear to be directly correlated with their sub-cellular localisation, for example TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10 recognise components of bacterial and fungal cell walls and are therefore largely present on the cell surface, whereas TLR3, TLR7, TLR8 and TLR9 recognise viral and bacterial nucleic acids in intracellular vacuoles [Citation10]. Ligands recognised at the cell surface include lipopolysaccharide (LPS) present on gram-negative bacteria (by TLR4), as well as gram-positive bacteria component peptidoglycan (by TLR2). TLR ligands detected within the cell are often virus- or prokaryote-derived nucleic acids, for example TLR3 recognises double-stranded (ds)RNA during viral infection, whereas TLR9 binds unmethylated CpG DNA from prokaryotes and DNA viruses [Citation11, 12].
Signaling through TLRs can induce inflammatory responses such as cytokine and chemokine expression, which as described previously helps orchestrate innate and adaptive immunity [Citation11, Citation13, Citation14]. TLR signaling often results in activation of NF-κB and MAP kinases (MAPKs), both are major components that regulate gene expression. NF-κB is a transcription factor that is sequestered in the cytoplasm of inactive cells by proteins such as inhibitor of NF-κB (IκB)α and IκBβ. Upon activation of TLRs, IκB proteins are phosphorylated by the trimeric complex IκB kinase (IKK), which contains the subunits: IKKα, IKKβ and IKKγ, and is regulated by multiple MAPK kinase kinases, TAK1 being the most studied. Phosphorylation of IκB proteins labels them for degradation by the proteasome; this releases NF-κB allowing its translocation into the nucleus.
A major adaptor upstream of these pathways is MyD88 that controls IκB phosphorylation and directly interacts with the TIR domain of the TLR [Citation9]. MyD88, TIRAP, TRIF, TRAM and SARM are all members of a family of cytoplasmic TIR domain-containing adaptor proteins that can interact with TLRs. It is these adaptor molecules that give signaling specificity between TLR members, although MyD88 is able to interact with almost all TLRs (with the exception of TLR3) as well as the other adapter proteins mentioned [Citation9, Citation15]. As a result of this, TLR signaling is often viewed as either a MyD88-dependent or independent process. TLR4 displays the signaling specificity gained from using multiple TIR domain-containing adaptors in MyD88-independent pathways; however, it can also activate MyD88-dependent pathways recruiting TIRAP to induce proinflammatory cytokine expression. In the case of other TLRs, MyD88-dependent pathways induce proinflammatory cytokine and type I IFN production, whereas MyD88-independent pathways involving TLR3 and TLR4, TRIF and TRAM are used to activate TRIF-dependent pathways leading to expression of type I IFN [Citation9].
NOD-Like Receptors
NLRs are a family of intracellular PRRs that play important roles in innate immune response upon recognition of PAMPs within the cytoplasm. In addition, they are also believed to be involved in both pro- and anti-inflammatory responses, and are therefore key modulators of the adaptive immune response [Citation16]. So far, 23 human and 34 murine NLRs have been discovered, of which both are made up of two subfamilies characterised by different N-terminal domains [Citation14]. The Nod subfamily members contain at least one caspase-activating and recruiting domain (CARD), whereas the NLRP subfamily members are defined by the presence of a pyrin domain. These domains allow the activated receptor to interact with downstream adaptor proteins in a homotypic CARD–CARD or pyrin–pyrin formation. Aside from these domains, NOD-like receptors have similar structures; they contain a C-terminal domain involved in direct or indirect binding with PAMPs, as well as a central NOD (alternatively NACHT) domain involved in oligomerisation with other activated NLRs [Citation17, 18].
Notable members of the Nod subfamily are NOD1 and NOD2, and are among the most well-studied receptors within the NLR family. They are known to recognise PAMPs found on the surface of bacteria, specifically, NOD1 recognises gram-negative bacteria peptidoglycan component, meso-diaminopimelic acid, whereas NOD2 recognises muramyl dipeptide, a component of almost all bacteria, with a few exceptions [Citation18]. NOD1 and NOD2 contain one and two CARDs, respectively, and upon activation by a ligand, they associate with the CARD-containing signaling adapter RIP2 (alternatively RICK) in a CARD–CARD manner. Active RIP2 is able to remove inhibition of NF-κB through ubiquitination of IκBγ, facilitated by the E3 ubiquitin ligases, cIAP1 and cIAP2 [Citation16]. NOD2 has been at the centre of many genome-wide studies that have revealed 30–40 polymorphisms within the NOD2 sequence associated with Crohn's disease, an inflammatory disease that affects the intestinal tract. It is widely agreed that Crohn's disease is caused by an increase in the inflammatory activity against normal gut microflora, and it is currently hypothesised that NOD2 polymorphisms lead to dysfunctional regulation of inflammation-inducing gut bacteria [Citation19]. Unlike NOD1 and NOD2, other members of the Nod family such as NLRC3 and NLRC5 are understudied. Evidence thus far points towards an inhibitory role for NLRC3, as over-expression reduces MyD88-dependent NF-κB production via the signaling intermediate, TRAF6 [Citation16, Citation20], whereas NLRC5 has only been recently discovered and has yet to have a determined role within innate immunity [Citation21].
The NLRP subfamily contains receptors NLRP1–12, where protein interactions with downstream signaling molecules occur through a pyrin domain. The most intriguing property of certain members of this NLR subfamily is their ability to form a multimeric complex known as the inflammasome. NLRP1, NLRP2, NLRP3, NLRC4, NLRC5, NLRP6 and NLRP7 all have the ability to form an inflammasome after ligand-induced activation [Citation16]. The inflammasome plays an important role in innate immunity, where the ligand-activated NLR associates with the non-NLR protein ASC along with pro-caspase-1 and forms the basic unit of the inflammasome. These units can interact in an assembly analogous to the apoptosome, forming a large multimeric protein complex. From this, the inflammasome cleaves pro-caspase-1 to caspase-1, which in-turn cleaves TLR-induced pro-IL-1β to IL-1β, an important regulator with the ability to direct local or systemic immune response. As well as IL-1β, the inflammasome also aids the maturation of IL-18 [Citation14]. Of all inflammasome complexes, the NLRP3 inflammasome is the most extensively studied. The NLRP3 inflammasome is known to be directly and indirectly activated by multiple molecules including PAMPs found on bacteria and fungi, endogenous ligands such as palmitate and ceramide, and non-biological environmental crystals such as uric acid, silica and asbestos [Citation14, Citation22, Citation23]. There is potential cross-talk between NLRP3 and Spleen tyrosine kinase (Syk)-activating receptors including the C-type lectin-like receptors discussed later, which has important implications in anti-microbial immunity.
RIG-I-Like Receptors
There are currently three known RLRs: RIG-I, MDA5 and LGP2. These receptors act as cytoplasmic surveillance proteins that detect viral RNA that is either directly inserted into host cells, or released from viral capsules after receptor mediated uptake and degradation. Structure of RLRs contains a ligand-binding domain for recognition of dsRNA and, with the exception of LGP2, tandem CARDs at the N-terminus. RIG-I also contains a repressor domain involved in controlling RIG-I-induced signaling [Citation14, Citation24]. In terms of ligand binding, RIG-I and MDA5 can recognise RNA viruses; however, each respond differently depending on the species of virus. Paradoxically, LGP2 acts as a positive regulator of both RIG-I and MDA5, even though it lacks a signal-transducing domain; however, it is thought that LGP2 acts upstream of these receptors to aid in recognition of the virus [Citation14, Citation25].
Signaling through RLRs leads to recognisable outcomes of innate immune response such as NF-κB activation; however, activation pathways differ from those previously described for other PRRs. In the case of active RIG-I and MDA5, interferon-β promoter stimulator-1 (IPS-1, alternatively MAVS) is recruited to the CARD domain and activates NF-κB via caspase-8, caspase-10, TRADD and FADD [Citation14, Citation26, Citation27]. As well as this, IPS-1 is used as an RLR signaling adaptor to induce and stabilise antiviral response via two intracellular organelles: mitochondria and peroxisomes. Upon RLR-dependent activation, IPS-1 can localise to the peroxisome inducing IRF1 activation that results in a fast-acting response to infection. IPS-1 can also localise to mitochondria where delayed, yet lasting response to viral infection is induced through activation of IRF3 and type I interferons. These allow the generation of potent responses to viral invasion [Citation14, Citation28].
C-TYPE LECTIN-LIKE RECEPTORS
C-type lectins (CTLs) were originally defined as a family of soluble and transmembrane receptors that bound carbohydrates via a carbohydrate recognition domain (CRD); however, continued research has revealed many structurally homologous domains to the CRD that are not restricted to carbohydrate binding. These domains are known as C-type lectin-like domains (CTLDs) and can be found on C-type lectin-like receptors (CTLRs) in varying numbers [Citation29].
Association between the CRD and carbohydrate ligand is facilitated by amino acid motifs with the ability to bind either mannose- or galactose-based structures. This interaction involves hydrogen bonding between a single calcium ion and carbonyl groups present at specific loci in the CRD fold; the calcium ion is also involved in hydrogen bonding with the carbohydrate itself. There are four sites where calcium coordination takes place; most important of these is site 2 as it supplies most of the binding affinity for the associated carbohydrate. The type of carbohydrate recognised depends on the amino acid within which the carbonyl group resides. For example, galactose binding is dependent on the following sequence, Gln-Pro-Asp, whereas mannose recognition requires a slightly modified sequence, Glu-Pro-Asn [Citation30, 31]. Despite loss of these residues, as well as residues important for calcium coordination, certain CTLDs can recognise a more diverse set of ligands including lipids and proteins [Citation32]. This group of CTLs are now referred to as CTLRs, and are of particular interest here.
CTLRs are a family of protein receptors crucial in the recognition of self- and non-self-ligands. They are so-called due to the presence of one or more CTLDs, and the 1000+ members are organised into 17 groups depending on their phylogeny and domain organisation [Citation30]. Ligands bind to the structurally conserved CTLD, which is observed to be a tight-fold constructed from loops stabilised by two disulphide bridges [Citation31]. Upon ligand binding to this fold, CTLRs can mediate a diverse set of downstream responses including, for example uptake of microorganisms, regulation of homeostasis such as clearance of apoptotic cells, direction of the adaptive immune response through cytokine and chemokine expression, and cell–cell adhesion [Citation31, Citation33].
Receptors of particular significance in this review are the myeloid CTLRs and their close relatives. These receptors share similar downstream pathways yet their physiological effects are often unrelated. Structural categorisation aside, myeloid CTLRs can be divided into three main groups depending on their intracellular signaling motifs: immunoreceptor tyrosine-based activatory motif (ITAM)-bearing, immunoreceptor tyrosine-based inhibitory motif (ITIM)-bearing and non-immunoreceptor tyrosine-based motif-bearing receptors [Citation31]. CTLRs can signal directly, through integral motifs in their cytoplasmic tails, or indirectly, through association with adaptor chains.
Most activation receptors associate with ITAM-bearing adaptor chains such as FcRγ chain, including DC-associated C-type lectin (Dectin)-2 and macrophage-inducible C-type lectin (Mincle), and DAP12 that can associate with myeloid DAP12-associating lectin (MDL)-1, for example. The ITAM consensus motif sequence is [YxxI/Lx(6–12)YxxI/L] and activation occurs when ligand binding causes receptor clustering and phosphorylation of tyrosine residues within this ITAM sequence by a member of the Src family kinases. Many ITAM-associated myeloid receptors utilise the kinase, Syk, where both SH2 domains of Syk interact with the two phosphotyrosines within the ITAM to render Syk fully active [Citation34].
Signaling pathways downstream of Syk activation form a large network of which the ultimate outcome is modulation of innate and adaptive immune responses. Pathways include activation of MAPK leading to large-scale changes in transcription of multiple genes, reorganisation of the actin cytoskeleton through activation of the Rac and Rho GTPase enzymes and activation of NF-κB through multiple routes. A well-understood area of CTLR signaling involves phosphoinositide 3-kinase (PI3K) generating membrane-associated phosphatidylinositol-3,4,5-triphosphate (PIP3) to activate its most noteworthy downstream enzyme, phospholipase Cγ2 (PLCγ2). Induction of the inositol triphosphate (IP3) and diacylglycerol (DAG) pathway through PLCγ2 activation leads to calcium ion release as a second messenger and activation of various protein kinase C (PKC) isoforms [Citation35]. PKC can activate relatives of the CARD-containing adaptor, CARD9, which forms a trimeric complex with Bcl-10 and Malt1. Exact mechanisms linking PKC directly to the CARD9-containing complex, rather than CARD9-related proteins, remain a mystery; however, it is known that CARD9-deficient cells do not activate NF-κB and subsequently lack cytokine and chemokine responses, suggesting its integral role in activation of NF-κB in a Syk-dependent pathway [Citation36].
Intriguingly, the presence of altered ITAM motifs, termed ITAM-like (alternatively hemITAM) motifs, appears to activate Syk-dependent pathways even though only a single tyrosine is present within this motif. Whereas the canonical ITAM motif contains a repeat of YxxI/L, an ITAM-like motif is often described as just a single YxxI/L. At first, this appears inconsistent with current knowledge of Syk signaling and its requirement for two individually phosphorylated tyrosines; however, the current hypothesis is that Syk is able to bridge two receptors that contain a single phosphotyrosine [Citation4].
Downstream signaling from ITAM/ITAM-like motifs are not restricted to the CARD9-containing complex; in the case of Dectin-1 (an ITAM-like-containing receptor) and Dectin-2 (an FcRγ-associated receptor), the CARD9 pathway is one of many transduction pathways initiated by the ITAM motif. For instance, NFAT activation through Dectin-1 results in characteristic cytokine patterns such as increased IL-2 and IL-10 in DCs. ERK and JNK MAPK pathways are also associated with NFAT activation, as both require the active PLCγ2 pathway for continuous transduction. In addition, Dectin-1 and Dectin-2 are also both associated with production of ROS (discussed earlier), which has implications in the secretion of IL-1β by aiding activation of the NLRP3 inflammasome [Citation22, Citation31].
As discussed, ITAM signaling is very versatile and these pathways can lead to physiological effects such as phagocytosis, cell proliferation, migration, differentiation and adhesion. However, activation of these responses is only half of the story, controlling these mechanisms is also a vital process. Receptors containing ITIM motifs are often implicated in negative regulation of pathways activate through ITAMs.
The ITIM sequence is described as [I/V/L/SxYxxI/L/V], where the central tyrosine is phosphorylated by the Src family kinases. However, rather than recruiting Syk, ITIM-bearing receptors recruit tyrosine phosphatases to negatively regulate the activity of signaling pathways [Citation4]. In many cases, ITIM-containing CTLRs associate with the SH2-domain-containing protein tyrosine phosphatase (SHP)-1 and SHP-2, which are able to directly affect the signaling capabilities of JAK/STAT and ITAM pathways. SHP-1 and SHP-2 are also able to regulate TLR pathways reducing NF-κB activity; however, these interactions are not yet fully understood, and could potentially involve reduction of synergistic collaboration between TLRs and other PRRs [Citation31]. As well as SHP-1 and SHP-2, association can occur between an ITIM motif and SH2 domain-containing inositol phosphatases (SHIP)-1 and SHIP-2, inhibiting signals that are initiated by PI3K through dephosphorylation [Citation37].
There are also CLRs and CTLRs that signal through motifs that do not fit the recognised ITAM, ITAM-like or ITIM sequences. Signaling pathways involving such motifs are unknown for most; however, for a few receptors, some pathways have been somewhat illuminated. One such elucidated receptor is DC-SIGN; it contains two of the most notable non-immunoreceptor tyrosine-based motifs, the di-leucine motif and the tri-acidic motif that are thought to be involved in modulation of other receptor pathways including the TLR signaling [Citation38]. Specifically, these motifs appear to be able to modulate Raf-1 activity, a MAPK kinase kinase, leading to the phosphorylation and subsequent recruitment of histone acetyltransferases (HATs) to p65, a subunit of NF-κB. Recruitment of HATs to p65 results in its acetylation and subsequently revealing promoters for IL-6, IL-10 and IL-12, increasing their transcription. DC-SIGN is not the only receptor to activate Raf-1 in a tyrosine-independent manner but Dectin-1 has also been implicated in this although the mechanisms involved are unclear [Citation31, Citation36]. It is important to note that these responses occur alongside TLR pathways after TLR-specific activation, as a form of signal modification [Citation39, 40]. It is also apparent that these motifs may act as a form of pathogen differentiation, for example formation of a multiprotein complex known as the signalosome in DC-SIGN signaling is highly dependent on the activating ligand present; specifically mannose-based ligands recognisable by DC-SIGN retain the signalosome form, allowing activation of downstream effectors, whereas recognition of fucose-based ligands causes dissociation of this complex [Citation39]. Other receptors utilising unusual pathways include murine SIGNR1 that signals through non-immunoreceptor tyrosine-based motifs and shows collaboration with TLRs and Dectin-1. Evidence for this is that ROS production in response to Candida albicans through Dectin-1 is enhanced by SIGNR1 [Citation41].
The Dectin-1 cluster is one of many receptor clusters that reside within the natural killer gene complex (NKC) whose functions span many areas of immunity and homeostasis. Many of the signaling motifs discussed for CTLRs above can be found in cytoplasmic tails of Dectin-1 cluster receptors and these CTLR have a broad range of functions. In the remainder of this review, we will discuss the receptors within this cluster in detail, including their ligands, signaling pathways, synergistic collaborations and their roles in immunity and homeostasis.
THE DECTIN-1 CLUSTER
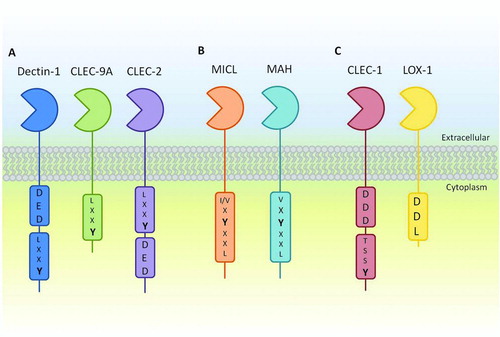
The Dectin-1 cluster form part of the group V C-type lectin-like receptors that have a single CTLD connected to an intracellular signaling domain via a stalk and transmembrane region and were thought to have arisen through gene duplication. These receptors lacking carbohydrate-binding motifs and largely recognise ligands in a calcium-independent manner. Receptors forming the Dectin-1 cluster include CLEC-1, CLEC-2, Dectin-1, CLEC-9A, Myeloid Inhibitory C-type-like Lectin (MICL/CLEC-12A), Macrophage Antigen H (MAH/CLEC-12B) and Lectin-like Oxidised LDL receptor-1 (LOX-1) (). Each one has the ability to regulate its own downstream signaling, analogous to that of the TLRs, due to the presence of one or more functional motifs within their intracellular tails. These receptors can be found primarily, but not entirely, on myeloid cells such as DCs, macrophages and neutrophils, where they can orchestrate all of the functions described for CTLRs earlier. Functions of receptors within the Dectin-1 cluster are predetermined, in that signaling motif(s) present within their intracellular domain dictate how they respond to extracellular stimuli, these motifs are ITAM-like, ITIM and tri-acidic sequences [Citation31, Citation33, Citation42].
FIGURE 1. Schematic representation of the family of group V type II CTLRs known as the Dectin-1 cluster. A: The activatory receptors Dectin-1, CLEC-2 and CLEC-9A containing ITAM-like and, with the exception of CLEC-9A, tri-acidic motifs important for downstream signaling. B: The inhibitory receptors MICL and MAH containing canonical ITIM motifs. C: Receptors containing novel motifs including C-type lectin-like scavenger receptor LOX-1, containing a DDL motif and CLEC-1 containing an uncharacterised tyrosine-based motif as well as a tri-acidic motif.
Dectin-1 (CLEC-7A)
Dectin-1 is a CTLR that was discovered to bind β-1,3-glucans and mycobacteria as well as an endogenous ligand present on T-cells [Citation43, 44]. Since then, Dectin-1 has become one of the most intensively studied CTLRs with many of its characteristics well defined, and it has becoming a model receptor for signaling CTLRs. Although its name originates from “dendritic-cell-associated C-type lectin 1”, Dectin-1 is not exclusively expressed on DCs and can be found on other cell types including macrophages, neutrophils and monocytes.
Like other group V type II transmembrane receptors, Dectin-1 contains a single extracellular CTLD, which is involved in calcium-independent ligand interactions, connected to a single-pass transmembrane domain by a stalk region. Attached to the cytoplasmic end of the transmembrane region lays a small intracellular tail, where the Dectin-1 ITAM-like [YxxL] and tri-acidic [DED] motifs reside [Citation33]. Both murine and human Dectin-1 homologues have multiple splice variants although each has two major isoforms. The major isoforms differ in either the gain or lack of the stalk, isoforms A and B, respectively, and are the only receptors capable of binding β-glucans [Citation4]. Dectin-1 is also N-glycosylated, affecting its expression and function [Citation33].
Through the recognition of β-glucans, Dectin-1 binds several fungal species such as Aspergillus, Candida, Coccidioides, Penicillium, Pneumocystis and Saccharomyces and because of this, focus on the immunological role for Dectin-1 has been directed at fungal immunity. Upon binding of β-glucans, Dectin-1 can induce a vast array of cellular effects including actin-mediated phagocytosis, activation of the respiratory burst through production of ROS, endocytosis, DC maturation and changes in cytokine and chemokine expression patterns such as TNF-α, IL-1α, IL-1β, IL-6, CXCL2, CCL3 and GM-CSF [Citation5, Citation33]. Although recent data suggest that Dectin-1 does not play a role in controlling C. albicans colonisation of the GI tract, Dectin-1 does play a major role in systemic candidiasis and other mucosal infections. Dectin-1 can direct effective antifungal mechanisms through T-helper cell (TH)-1 and 17 responses, which are defective in human patients with homozygous non-functional Dectin-1 [Citation45, 46]. In addition to fungal ligands, Dectin-1 can also recognise an unidentified ligand on mycobacteria, which leads to the production of IL-12 [Citation31].
Dectin-1 signaling downstream of ligand binding involves both Syk-dependent and Syk-independent signaling cascades (), along with pathways from collaborating receptors such as those of TLRs. Within these networks, pathways leading to the Syk-dependent activation of NF-κB can be categorised into both canonical and non-canonical routes. The canonical pathway is as described for the TLRs, where the phosphorylation of IκB complex by IKK releases NF-κB allowing its translocation into the nucleus where it can regulate gene transcription. However, unlike TLRs, Dectin-1 activates this IKK pathway through Syk association with an ITAM-like motif, where active Syk can phosphorylate PLCγ2, allowing subsequent activation of the CARD9-Bcl10-Malt1 complex via PKCδ.
FIGURE 2. Signaling network of Dectin-1. Multiple pathways downstream of Dectin-1 rely on Syk. This can lead to PLCγ2/Ca2+ signaling leading to PKCδ activation that controls calcineurin/NFAT and ROS/NLRP3 inflammasome, and the phosphorylation of CARD9-Bcl10-Malt1. The CARD9 complex is able to activate all NF-κB subunits including c-Rel by removing its inhibitor, IκB, through activation of the IKK complex. Syk can also activate IKK complex through the non-canonical NIK pathway, leading to the activation of NF-κB subunits RelB and p52. This pathway can be inhibited the Raf-1-mediated pathway involving p65. Phosphorylated and acetylated p65 can interact with RelB, sequestering it away from an active NF-κB complex. Resultant activation of NFAT and NF-κB results in modification of gene transcription including cytokines and chemokines. Dashed arrows represent a pathway that has yet to be fully defined.
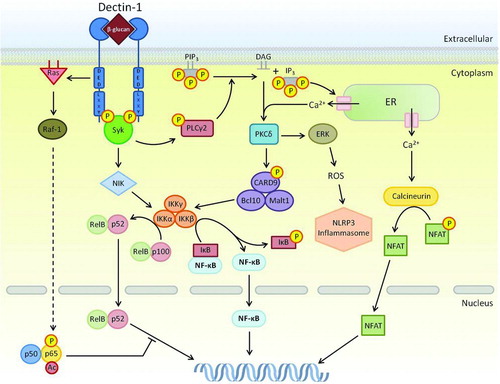
PLCγ2 is known to indirectly interact with a close relative of CARD9 called CARMA1 in other receptor pathways. This interaction is based on the hydrolytic activity of PLCγ2 on the phosphorylated membrane phospholipid PIP3 and results in membrane-anchored DAG and soluble IP3, both of which aid in activation of PKC isoforms. Active PKC can stimulate CARMA1, allowing formation of a complex with Bcl-10 and Malt1, much like the CARD9 complex present in Dectin-1 signaling [Citation47]. Recent evidence has shown that the CARD9 activation pathway is analogous to the CARMA1 pathway, as CARD9 phosphorylation is heavily reliant upon the PKCδ isoform in response to Dectin-1 stimulation. Indeed, PKCδ knockout DCs are defective in Dectin-1-mediated responses. The CARD9-Bcl10-Malt1 complex in Dectin-1 signaling is known to activate subunits of NF-κB through the IKK complex as described for the TLRs, and of particular note is Malt1-dependent activation of the subunit c-Rel, which is required for TH17-inducing cytokines IL-1β, and IL-23p19, and immunity against Candida infection [Citation32, Citation48, Citation49].
Aside from the canonical NF-κB activation pathway, Dectin-1 also mediates a non-canonical pathway utilising NF-κB-inducing kinase (NIK) through Syk activation. NIK can activate IKKα leading to production of p52 from its precursor, p100. Dimers of p52 and RelB can translocate into the nucleus and induce transcription [Citation32]. In addition, Dectin-1 activates a Syk-independent pathway through its tri-acidic motif [DED] involving Raf-1 activation by Ras, the end result of which is a merging of non-canonical NF-κB and Raf-1 pathways. Raf-1-induced phosphorylation of p65 results in the formation of a p65-RelB dimer that sequesters active RelB, this prevents RelB binding to DNA, and favours Raf-1-dependent IL-12p70 production required for TH1 differentiation, as well as IL-1β, IL-23, IL-6 that are involved in TH17-polarising cytokines [Citation4, Citation31, Citation32, Citation49]. The DED motif is also required for efficient phagocytosis by this receptor [Citation50].
This network of Dectin-1-induced pathways also involves the activation of the transcription factor, nuclear factor of activated T cells (NFAT). Dectin-1 signals through the classic calcineurin/NFAT pathway through Syk activation of PLCγ leading to soluble IP3 as described earlier. IP3 is able to bind endoplasmic Ca2+ channels, resulting in an influx of Ca2+ into the cytoplasm. This increase in calcium concentration induces calcineurin activation and consequently, dephosphorylation of NFAT and translocation into the nucleus. NFATs role within the innate immune system, in particular DC cytokine expression, is potent expression of IL-2 and IL-10, regulating development in lymphocytes and Tregs, as well as differentiation and survival of the latter [Citation51].
It is now widely accepted that receptors collaborate and that this is an important component for effectively controlling infection. Indeed Dectin-1 can collaborate with many MyD88-coupled TLRs including TLR2, TLR4, TLR5, TLR7 and TLR9 [Citation4]. Coupling between Dectin-1 and TLR2 has been shown to be important in phagocytosis as well as MAPK activity in the control of Candida infections, where cooperative signaling is required to trigger effective cytokine production and drive TH17 responses [Citation52]. Furthermore, Dectin-1 has been shown to collaborate with SIGNR1 inducing TNF [Citation53], TLRs including TLR2, TLR4, TLR5, TLR7 and TLR9 through MyD88 and Syk-dependent pathways [Citation54] as well as the tetraspanin CD63, during phagocytosis of Saccharomyces cerevisiae in DCs [Citation55]. Dectin-1 is also able to activate complement receptor 3 (CR3, alternatively Mac-1) to enhance fungal recognition and neutrophil response through collaborative function. This involves the activation of downstream Vav1 and Vav3 as well as PLCγ by Dectin-1 to activate CR3, resulting in combined signaling to enhance phagocytosis and ROS production of neutrophils at the site of infection [Citation56].
CLEC-9A
CLEC-9A is structurally similar to other group V type II transmembrane receptors described, and contains an ITAM-like motif [YxxL] within its intracellular tail. There are multiple splice variants of murine CLEC-9A; the 6-exon variant is expressed as a non-glycosylated monomer, whereas the 7-exon variant is expressed as a dimer. This variant was shown to be expressed on plasmacytoid DCs and, interestingly, CD8α+ DCs that are highly efficient at antigen cross-presentation [Citation57, 58]. Human CLEC-9A is predominantly expressed as glycosylated dimers on BDCA3+ DCs that are known to be the equivalent to murine CD8α+ DCs due to similarities in expressed surface markers [Citation59]. The extracellular region of CLEC-9A contains a Ca2+-independent CTLD that binds a proteinaceous ligand known to be ubiquitous to all cell types and exposed during necrotic cell death. Recent developments have uncovered this endogenous ligand to be F-actin; a major, highly conserved component of the cytoskeleton from yeast to man [Citation60, 61]. CLEC-9A has been shown to help mediate cross-presentation of necrotic cell-associated material for priming of cytotoxic T cells, acting as a link between innate recognition and T-cell responses [Citation61, 62]. Interestingly, recent findings have suggested that CLEC-9A-dependent cross-presentation involves endocytosed material that is presented via the internal cross-presentation pathway, rather than the external pathway [Citation63]. However, unlike Dectin-1, binding of F-actin to CLEC-9A had no appreciable effect on cellular cytokine responses suggesting that CLEC-9A is not an activation receptor and that cross-presentation is its primary function [Citation61, Citation64]. Additional evidence for its non-activation function was shown through antibody cross-linking of CLEC-9A, which also had no effect on DC activation; likewise, Dectin-1/CLEC-9A chimeric receptors stimulated with β-glucans showed no signs of DC activation [Citation31, Citation57, Citation58]. On the other hand, mutation of an isoleucine to a glycine directly upstream of the ITAM-like motif within the Dectin-1/CLEC-9A chimera results in increased cytokine production, suggesting that CLEC-9A can activate myeloid cells, but that this function is restricted by the presence of an isoleucine within the cytoplasmic tail [Citation63].
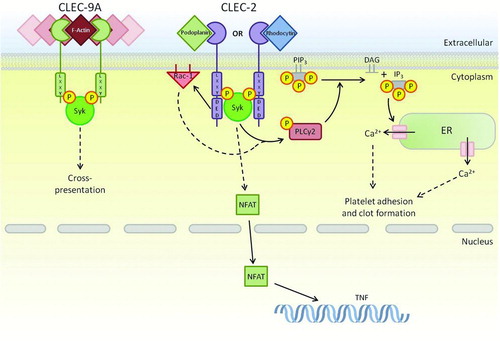
Linking CLEC-9A functions to signaling networks is in its infancy; however, what is known is that cross-presentation is dependent on Syk recruitment to the phosphorylated ITAM-like motif (). Investigations into the ITAM-like motif revealed that antibody cross-linking to the CTLD induced tyrosine phosphorylation and subsequent Syk recruitment in vitro, this was also observed when CLEC-9A was exposed to necrotic cells in vitro. Continued research on the role of CLEC-9 as ITAM-like motif used tyrosine mutant receptors to show that the tyrosine was required for functional cross-presentation. These mutants showed marked reduction in cross-presentation [Citation65].
FIGURE 3. ITAM-like-bearing CTLRs within the Dectin-1 cluster. As well as Dectin-1, there are two additional receptors that contain ITAM-like motifs. These receptors share Syk as a primary downstream signaling kinase; however, the lack of a tri-acidic in CLEC-9A and the presence of an isoleucine prevent the endocytic and activation functions of this receptor. CLEC-2 utilises its tri-acidic motif to complement activation through Syk resulting in adhesion and clot formation in platelets. Dashed arrows represent a pathway that has yet to be fully defined.
CLEC-2 (CLEC-1B)
CLEC-2 was discovered along with CLEC-1 in a bioinformatic screen for NK receptors. Expression of CLEC-2 was determined by RT-PCR, where mRNA was found in bone marrow cells, DCs, monocytes, granulocytes and some NK cell populations; however, most studies have focussed on the role of CLEC-2 on platelets, where the receptor is expressed at the cell surface [Citation4]. Murine CLEC-2 has three splice variants, two of which are retained within the cytoplasm due to the lack of a transmembrane region. The sequence analysis revealed an ITAM-like motif [YxxL] present in the cytoplasmic tail similar to those within Dectin-1 and CLEC-9A and multiple studies have shown this to play an important role in platelet signaling in response to endogenous and exogenous ligands [Citation66, 67].
Known ligands of CLEC-2 include the endogenous protein podoplanin, a transmembrane sialoglycoprotein found in multiple cell types such as lymphatic endothelial cells, type I lung alveolar cells and some cancer cells. Podoplanin-deficient and CLEC-2-deficient mice have defects in vascular and lymphatic connections, leading to blood in the lymph [Citation31, Citation68]. HIV-1 is thought to be recognised by CLEC-2; however, it is likely that recognition involves a host protein that is incorporated during HIV-1 budding, as binding occurred in the absence of envelop protein. In the case of CLEC-2 recognition, this host protein has been hypothesised to be podoplanin as evidence suggests that HIV-1 can infect renal cells, a common site for podoplanin expression [Citation4].
CLEC-2 also binds the exogenous ligand rhodocytin, a multimeric protein found in snake venom that has been shown to lead to platelet activation, aggregation and subsequent coagulation of the blood [Citation67, Citation69]. Its tetrameric structure is thought to aid in CLEC-2 clustering at the cells surface resulting in amplification of downstream signaling [Citation4]. Binding of these two ligands may occur through different mechanisms as rhodocytin interacts directly with CLEC-2 [Citation70], whereas glycosylation of podoplanin is involved in CLEC-2 binding [Citation71].
The functions of CLEC-2 have become more apparent in recent years, for example platelet activation is now known to be an important factor in tumour metastasis, which may be triggered by up-regulation of podoplanin at the cell surface [Citation72]. In addition to this, CLEC-2 has also been implicated in arteriolar rate of flow in mice [Citation73] as well as phagocytic roles in neutrophils, suggesting a potential role within innate immunity [Citation74]. As with other CTLRs in the Dectin-1 cluster, signaling from CLEC-2 requires Syk association (), however, unlike knowledge currently described; this ITAM-like motif has different modes of activation compared to the canonical ITAM motif [Citation68, 69, Citation75]. Because of this, CLEC-2 has become somewhat of a model for ITAM-like signaling, with multiple features of this motif revealed in recent studies. Of particular interest here is the use of a tri-acidic motif [DED] present upstream of the ITAM-like motif that is able to facilitate tyrosine-based signaling and partake in ITAM-like-independent signaling [Citation68].
Mutational analysis of the CLEC-2 tri-acidic cluster showed that phosphorylation of the ITAM-like motif was markedly reduced and resulted in a complete loss of CLEC-2 signaling, suggesting that the tri-acidic motif is required for phosphorylation the ITAM-like motif. Another interesting observation is that in the presence of a highly selective Syk inhibitor, CLEC-2 ITAM-like phosphorylation was completely blocked, whereas phosphorylation of ITAM-containing adaptors such as FcRγ was not perturbed. In addition to this, phosphorylation of Syk was shown to precede phosphorylation of the ITAM-like motif in an Src-dependent manner. This suggests that CLEC-2 ITAM-like phosphorylation is dependent upon Syk, quite opposite to previous models, where Src family kinases phosphorylate the ITAM-like motif allowing Syk binding [Citation68, Citation75].
Functional studies into the phagocytic potential of myeloid-expressed CLEC-2 were done using chimeric receptors containing the extracellular and transmembrane domain of Dectin-1 fused to the signaling tail of CLEC-2. It was determined that CLEC-2 can induce phagocytosis, NFAT activation and TNF production; however, the receptor is unable to activate the respiratory burst. Although the mechanisms behind the Syk involvement in CLEC-2-mediated phagocytosis are still not fully understood, it is likely that the tri-acidic cluster upstream of the tyrosine is critical for this, as it is for Dectin-1. With multiple characteristics similar to Dectin-1, CLEC-2 may be able to function as a PRR in addition to its well-known homeostatic role in platelet activation [Citation74]. However, exogenous ligands (other than rhodocytin) are currently unknown.
Recent evidence suggests that phosphorylation of CLEC-2 is dependent on multiple factors; these include association with lipid rafts, actin polymerisation and Rac-1 activation [Citation76]. CLEC-2 was found within lipid rafts, presumably bringing it into close proximity with important downstream signaling components. This process was determined to be actin-independent; however, in the presence of an actin polymerisation inhibitor, CLEC-2 phosphorylation was reduced, suggesting that actin may have a role in Src function or recruitment [Citation73]. Furthermore, an increase in CLEC-2 ITAM-like tyrosine phosphorylation in the presence of the platelet secondary mediators ADP and Thromboxin A2 (TxA2) upstream of Rac-1 was observed. This increase in activation is able to bypass the need for actin polymerisation; therefore, Rac-1 activation may results in a feedback loop affecting the Src family kinases.
CLEC-2 is also thought to activate PLCγ2 via Syk-phosphorylation, along with additional support from active Rac-1 [Citation77]. PLCγ2, as discussed earlier, is a lipase that when activated in platelets leads to platelet-specific responses such as adhesion and clot formation through Ca2+ influx into the cytoplasm. Although the interplay between PLCγ2 and Rac-1 is not fully understood within CLEC-2 signaling, Rac-1 could be supporting PLCγ2 signaling through production of PLCγ2 substrates as well as directly interacting with it, as observed in other receptor pathways [Citation73].
Evidence of receptor co-stimulation also exists with CLEC-2 pathways, where addition of anti-CLEC-2 monoclonal (mAb) along with LPS can potentiate the effects of TLR function, further suggesting a role for CLEC-2 in inflammatory responses. This is supported by data that suggest CLEC-2 expression increases on myeloid cells after exposure to LPS, which also increases IL-10 production possibly through the Syk/NFAT activation pathway. A noteworthy point is that CLEC-2 activation was achieved using antibody crosslinking, and true ligands of this receptor may illicit different responses [Citation78].
MICL (CLEC-12A)
Human MICL (alternatively DCAL-2, CLL-1 and KLRL-1) was discovered in a genome search for sequences homologous to the CRD of DC-SIGN and DCIR. Unlike the previously discussed Dectin-1 cluster receptors, MICL is an inhibitory CTLR, containing an ITIM motif [human: VxYxxL, murine: IxYxxL] within its intracellular tail. There are at least three isoforms of human (h)MICL: α, β and γ, where β-isoform lacks the transmembrane domain and γ-isoforms lacks the CTLD. The receptor also contains cysteine residues that are often associated with dimerisation; however, hMICL is found as a monomer and is heavy glycosylated. This is unlike its murine counterpart that is expressed as a dimer and lacks heavy glycosylation.
MICL is principally expressed on myeloid cells including DCs and macrophages, and is down-regulated during stimulation with TLR ligands, cell migration into peripheral tissues and sites of inflammation; a classic response seen in many inhibitory receptors. Specific ligands for MICL are currently undefined; however, soluble MICL was used to probe for potential ligands in many different tissues revealing binding in bone marrow, thymus, heart, spleen and kidney. This widespread recognition suggests that MICL may play a role in regulation of homeostasis [Citation33, Citation79, Citation80]. MICL is also expressed on acute myeloid leukaemia (AML) cells, where it is used as a marker and having a potential role in AML therapeutics. Only 30–40% of patients affected by AML survive due to the tolerance this cell type has to chemotherapy upon relapse. Recent progress using MICL as a therapeutic target involved the development of cytotoxic anti-MICL mAbs to target AML-derived cells. These mAbs showed strong cytotoxicity against 94% of AML-derived cells, demonstrating a promising approach to the treatment of AML using MICL as a target [Citation81, 82].
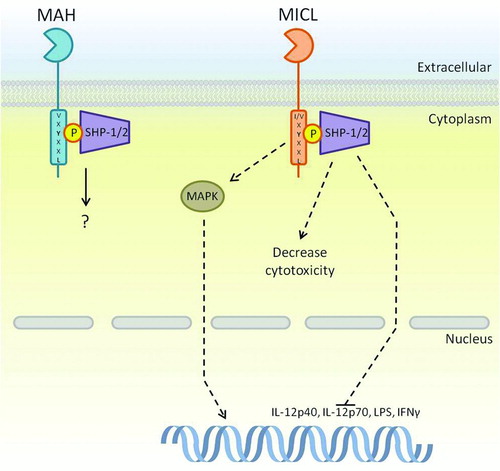
Of particular interest here is MICLs ability to regulate the activation responses downstream of PRRs including TLRs and CTLRs, as this would have direct effects on cellular response during pathogenic invasion. As described earlier for ITIMs, MICL recruits SHP-1 and SHP-2 via their SH2 domains to the phosphorylated tyrosine within the ITIM, and this pathway is thought to mediate the inhibitory function of this receptor (). MICL's inhibitory function has been directly demonstrated using a fusion of the Dectin-1 CTLD with the signaling tail of MICL. When this MICL/Dectin-1 chimeric receptor was co-expressed with full length Dectin-1, MICL signaling could successfully inhibit zymosan-induced stimulation of Dectin-1 [Citation80]. During another study, the effects of MICL on cytokine and chemokine production were investigated; MICL was shown to suppress zymosan and LPS-induced IL-12p40 and IL-12p70 as well as LPS-induced TNF expression [Citation83]. In addition to this, anti-MICL antibody cross-linking suppressed TLR-induced IFN-γ production in mature DCs; however, in DCs activated through CD40, IFN-γ production was activated, suggesting differential roles depending on the method of DC activation. MICL is also described to have an inhibitory role in NK cell cytotoxicity, where blocking of potential MICL ligands increased NK toxicity [Citation84]. Antibody cross-linking of MICL is also able to stimulate MAPK pathways in immature DCs (iDCs) and although these DCs do not mature, MICL was able to induce cytokine and chemokine expression as well as antibody internalisation. This suggests that in certain circumstances MICL may also have an activation role [Citation83], which has been described for other inhibitory receptors [Citation79].
FIGURE 4. ITIM-bearing CTLRs within the Dectin-1 cluster. The ITIM motif within MICL and MAH is known to interact with SHP-1 and SHP-2; however, pathways downstream of these phosphatases are only partially known. MICL is known to inhibit cytokine transcription instigated by other receptors as a method of control. On the other hand, there is evidence to suggest that MICL induces activatory pathways through MAPK, which is somewhat contradictory to the known functions of its motif. Downstream signaling of MAH is largely unknown. Dashed arrows represent a pathway that has yet to be fully defined.
MAH (CLEC-12B)
MAH was detected through computational analysis and has considerable homology to the activation receptor NKG2D. It has two isoforms, which are expressed differentially, one is expressed widely amongst cell types excluding the brain, whereas the other is expressed in tissues such as the lungs, mammary gland and ovaries; however, the latter is predicted to be non-functional due to the lack of a section of its CTLD. More recently, the presence of MAH mRNA was shown in DCs, monocytic cells and lymphocytes [Citation85]. Sequence data show that MAH contains an ITIM motif [VxYxxL] within its signaling tail, suggesting that it may well be an inhibitory partner of NKG2D; however, as they share no known ligands and their expression profiles on T cells and NK cells are different, this is unlikely [Citation86].
Only a few aspects of the human MAH signaling pathway are known (); it can associate with the tyrosine phosphatases SHP-1 and SHP-2 that can reduce the effects of kinases within ITAM-mediated pathways. Although MAH is most likely not an inhibitory partner of NKG2D, it is able to inhibit some pathways induced by NKG2D as well as other NK receptors including 2B4. Nevertheless, this inhibition was not enough to prevent NK cell activation [Citation86]. Knowledge of MAH function and physiological role will be greatly increased upon discovery of a ligand.
CLEC-1 (CLEC-1A)
CLEC-1 is almost completely uncharacterised and although there is evidence to suggest a few conserved residues between the mouse and humans, none of these have been proven to aid in downstream signaling to date. Bioinformatic predictions into the structure of CLEC-1 describe it as being a group V type II transmembrane receptor with some potentially noteworthy residues present within its intracellular tail [Citation66, Citation87]. Literature also describes two conserved cysteine residues within the stalk region that are hypothesised to allow CLEC-1 to form homodimers facilitated by disulphide bridges [Citation88]. However, functional work on CLEC-1 is restricted due to the lack of a known ligand. Notable residues within the intracellular tail are a tyrosine that presents itself in an undefined signaling sequence [YSST] as well as the presence of a tri-acidic cluster [DDD] that could potentially mediate signaling in ways analogous to tyrosine-independent signaling instigated by Dectin-1 [Citation66, Citation87] (). However there are no data to support either of these being a functional motif.
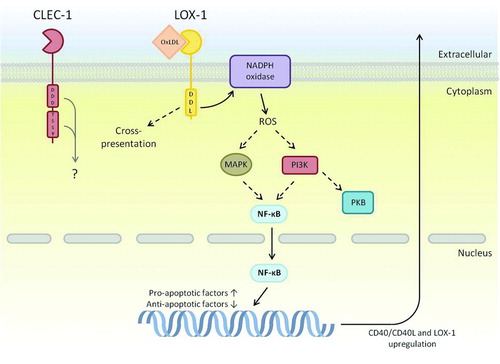
FIGURE 5. CTLRs within the Dectin-1 cluster that contain novel signaling motifs. Many components downstream of LOX-1s novel DDL motif have been determined, although exact mechanisms are not entirely identified. Interestingly, LOX-1 has the ability to up-regulate transcription of other receptors including itself; a mechanism that has been shown to aid in plaque formation during atherosclerosis. Downstream signaling of CLEC-1 is largely unknown; however, it does contain a tri-acidic motif that may have signaling potential due to similar signaling sequences in Dectin-1 and CLEC-2. Dashed arrows represent a pathway that has yet to be fully defined, and the grey arrow represents potential pathways based on other related receptor functions.
What is known of CLEC-1 is that it is expressed on myeloid and endothelial cells, and over-expressed in rat allograft tolerance studies, where its expression is up-regulated by IL-10 and TGFβ. Observations of function showed that alloantigen-induced CD4+ CD25+ T cells at the site of allograft increased expression of CLEC-1 in tolerated allograft endothelial cells. Interestingly, this up-regulation was not observed in rejected allografts, suggesting that CLEC-1 has a potentially protective role in transplantation. In addition to this, increased expression of CLEC-1 in tolerated allografts directly correlated with an increase in Foxp3 and a decrease in IL-17 expression, suggesting that CLEC-1 may regulate T-cell responses [Citation89].
LOX-1 (CLEC-8A)
LOX-1 was the first of the Dectin-1 cluster receptors to be discovered and is often considered a scavenger receptor, because of its ligands (see below). It is expressed in low levels primarily on endothelial cells as well as iDCs, macrophages, monocytes and B cells and induces signaling pathways within these cell types using a novel Asp-Asp-Leu (DDL) motif. Glycosylation of LOX-1 and its dimerisation at the cell surface is thought to aid ligand recognition, of which oxidised low-density lipoproteins (OxLDL) is the most well known. However, LOX-1 recognition is not restricted to OxLDL, it can recognise other oxidised lipid structures as well as HSP70 exposed in apoptotic cells, phospholipids, gram-positive and gram-negative bacteria, and activated platelets. Recognition of these ligands can result in a large variety of downstream responses such as antigen cross-presentation, endocytosis, phagocytosis, cytokine and NF-κB production, ROS production, cell adhesion and apoptosis, but the most noteworthy of LOX-1 functions is its role within vascular diseases including hypertension, cardiovascular disease and atherosclerosis [Citation90, 91].
LOX-1's role in vascular diseases has been intensively studied as it is often up-regulated in response to pro-atherogenic factors, leading to progression of atherosclerosis. Atherosclerosis is the formation of vascular plaques as a result of damage to the lining of the blood vessels often caused by oxidative stress from multiple risk factors including high blood pressure, smoking and diabetes mellitus. At the site of damage, LDL-cholesterol can enter into the sub-endothelial space becoming oxidised in the process, forming OxLDL. Being the primary cause of endothelial damage, OxLDL is ingested by scavenger receptors such as LOX-1, SR-A1 and SR-A2 present on the surface of macrophages, resulting in their conversion into foam cells and formation of a fibrous capsule to surround the damaged area, known as an atheroma [Citation92]. Multiple atheromas can cause blood vessel rigidity, increasing blood pressure and, therefore, causing further vessel damage in a positive feedback loop, leading to acute coronary syndrome (ACS). ACS is a major cause of mortality in developed countries; however, methods of detecting the coronary lesions typical of ACS do not exist. An important part of LOX-1 involvement in ACS is when it is cleaved to form a soluble receptor that circulates in the blood, a function that could be utilised as a biomarker for ACS. Unlike the biomarker used for cardiac muscle damage (troponin T), soluble LOX-1 concentrations in the blood peak earlier, therefore allowing the onset of ACS to be detected before plaques become damaged or rupture [Citation93–95].
Outside of vascular homeostasis, macrophages expressing LOX-1 have been recently shown to bind the GroEL homologue present on the surface of Escherichia coli. Not only were the macrophages able to bind E. coli, they were able to internalise the bacteria and increase LOX-1 expression upon binding. Interestingly, rather than aiding the removal of E. coli from the host, it made the host more susceptible to peritonitis. Pre-treatment of the host with an anti-LOX-1 antibody reduced susceptibility, suggesting that LOX-1 aids in the invasiveness of E. coli during infection [Citation96].
Multiple signaling pathways have been associated with LOX-1 upon activation with OxLDL; however, they are yet to be fully defined (). There is evidence to suggest that LOX-1 signaling involves nuclear translocation of NF-κB via activation of NADPH oxidase [Citation97]. NADPH oxidase induces the production of ROS, which is a second messenger that is able to activate MAPK and PI3K pathways, leading to NF-κB activation. In addition, LOX-1 activation is thought to up-regulate CD40 and CD40 ligand levels as well as up-regulation of monocyte chemoattractant protein-1 and adhesion molecules that aid in macrophage migration and infiltration of the exposed endothelial layer [Citation97, 98]. OxLDL-mediated activation of LOX-1 also results in apoptosis through activation of pro-apoptotic factors such as caspase-3 and caspase-9 as well as down-regulation of anti-apoptotic factors including Bcl-2 and cIAP1 [Citation99].
CONCLUSION
Receptors of the Dectin-1 cluster can be separated into two groups depending on their signaling functions: activation and inhibitory receptors. Although some of these receptors are largely uncharacterised, new and exciting functions are constantly being discovered, and not just in the recognition of pathogens but in the control of homeostasis and disease. Equally excitingly are the new insights into the pathways and mechanisms of intracellular signaling utilised by these receptors. Given the increasingly clear evidence of the importance of these receptors in immunity and homeostasis, gaining a better understanding of their functions and underlying mechanisms is essential if we are ever to utilise this knowledge for the development of novel immunotherapeutic approaches.
ACKNOWLEDGEMENTS
The authors thank the Wellcome Trust for funding.
Declaration of Interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
| ABBREVIATIONS | ||
| ACS | = | acute coronary syndrome |
| AML | = | acute myeloid leukaemia |
| ASC | = | apoptosis-associated speck-like protein containing a CARD |
| Bcl | = | B-cell lymphoma |
| BDCA | = | thrombomodulin/CD141 |
| CARD | = | caspase-activating and recruiting domain |
| CARMA | = | CARD recruited membrane-associated protein |
| CCL | = | chemokine ligand |
| CD | = | cluster of differentiation |
| cIAP | = | cellular inhibitor of apoptosis |
| CLEC | = | C-type lectin-like receptor |
| CR | = | complement receptor |
| CRD | = | carbohydrate recognition domain |
| CTL | = | C-type lectin |
| CTLD | = | C-type lectin-like domain |
| CTLR | = | C-type lectin-like receptor |
| CXCL | = | CXC ligand |
| DAG | = | diacylglycerol |
| DAP | = | DNAX-activation protein |
| DC | = | dendritic cell |
| DCIR | = | dendritic cell immunoreceptor |
| DC-SIGN | = | dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin |
| Dectin | = | dendritic cell-associated C-type lectin |
| FADD | = | Fas-associated protein with death domain |
| GI | = | gastrointestinal |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| HAT | = | histone acetyltransferases |
| HIV | = | human immunodeficiency virus |
| HSP | = | heat shock protein |
| iDC | = | immature DCs |
| IFN | = | interferon |
| IKK | = | inhibitor of NF-κB kinase |
| IL | = | interleukin |
| IL-1R | = | interleukin-1 receptor |
| IP3 | = | inositol triphosphate |
| IPS | = | interferon-β promoter stimulator 1 |
| IRS | = | interferon regulatory factor |
| ITAM | = | immunoreceptor tyrosine-based activatory motif |
| ITIM | = | immunoreceptor tyrosine-based inhibitory motif |
| IκB | = | inhibitor of NF-κB |
| JNK | = | c-Jun N-terminal kinases |
| LBD | = | ligand-binding domain |
| LDL | = | low-density lipoproteins |
| LOX | = | lectin-like oxidised LDL receptor |
| LPS | = | lipopolysaccharide |
| mAb | = | monoclonal antibody |
| MAH | = | macrophage antigen H |
| Malt | = | mucosa-associated lymphoid tissue lymphoma translocation |
| MAPK | = | mitogen-activated protein kinase |
| MDA | = | melanoma differentiation-associated protein |
| MDL | = | DAP12-associating lectin |
| MICL | = | myeloid inhibitory C-type-like lectin |
| Mincle | = | macrophage-inducible C-type lectin |
| MyD | = | myeloid differentiation primary response gene |
| NFAT | = | nuclear factor of activated T-cells |
| NF-κB | = | nuclear factor-κB |
| NIK | = | NF-κB-inducing kinase |
| NK | = | natural killer |
| NKC | = | natural killer gene complex |
| NKG2D | = | natural killer group 2 member D |
| NLR | = | nucleotide-oligomerisation domain-like receptor |
| NLRC | = | NOD, leucine-rich repeat and CARD domain-containing protein |
| NLRP | = | NOD, leucine-rich repeat and pyrin domain-containing protein |
| NOD | = | nucleotide-oligomerisation domain |
| OxLDL | = | oxidised low-density lipoproteins |
| PAMP | = | pathogen-associated molecular pattern |
| PI3K | = | phosphoinositide 3-kinase |
| PIP3 | = | phosphatidylinositol-3,4,5-triphosphate |
| PKC | = | protein kinase C |
| PLC | = | phospholipase C |
| PRR | = | pattern recognition receptor |
| RIG | = | retinoic acid-inducible gene |
| RIP | = | receptor-interacting serine/threonine-protein kinase |
| RLR | = | retinoic acid-inducible gene-like receptor |
| ROS | = | reactive oxygen species |
| RT-PCR | = | reverse transcriptase-polymerase chain reaction |
| SARM | = | sterile-alpha and armadillo-motif-containing protein |
| SH | = | Src homology |
| SHIP | = | SH2 domain-containing inositol phosphatases |
| SHP | = | SH2-domain-containing protein tyrosine phosphatase |
| SIGNR | = | SIGN-related |
| STAT | = | signal transducer and activator of transcription |
| Syk | = | spleen tyrosine kinase |
| TAK | = | transforming growth factor β-activated kinase |
| TGF | = | transforming growth factor |
| TH | = | T helper |
| TIR | = | Toll/IL-1 receptor |
| TIRAP | = | Toll-interleukin 1 receptor (TIR) domain-containing adaptor protein |
| TLR | = | Toll-like receptor |
| TNF | = | tumour necrosis factor |
| TRADD | = | tumour necrosis factor receptor type 1-associated death domain protein |
| TRAF | = | TNF receptor-associated factor |
| TRAM | = | TRIF-related adaptor molecule |
| Tregs | = | regulatory T cells |
| TRIF | = | TIR-domain-containing adapter-inducing interferon-β |
| TxA2 | = | thromboxin A2. |
REFERENCES
- Medzhitov R. Approaching the asymptote: 20 years later. Immunity 2009;30:766–775.
- Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell 2002;111:927–930.
- Janeway CA, Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989;54(Pt 1):1–13.
- Kerrigan AM, Brown GD. Syk-coupled C-type lectin receptors that mediate cellular activation via single tyrosine based activation motifs. Immunol Rev 2010;234:335–352.
- Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 2006;6:33–43.
- Kohchi C, Inagawa H, Nishizawa T, ROS and innate immunity. Anticancer Res 2009;29:817–821.
- Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol 2006;7:1029–1035.
- Leulier F, Lemaitre B. Toll-like receptors–taking an evolutionary approach. Nat Rev Genet 2008;9:165–178.
- Yamamoto M, Takeda K. Current views of Toll-like receptor signaling pathways. Gastroenterol Res Practice 2010;2010:240365.
- Tarang S, Kumar S, Batra SK. Mucins and Toll-like receptors: kith and kin in infection and cancer. Cancer Lett 2012;321:110–119.
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol 2005;560:11–18.
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004;5:987–995.
- Lamkanfi M, Kanneganti TD. Regulation of immune pathways by the NOD-like receptor NLRC5. Immunobiology 2012;217:13–16.
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011;30:16–34.
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801.
- Schneider M, Zimmermann AG, Roberts RA, The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol 2012;13:823–831.
- Rubino SJ, Selvanantham T, Girardin SE, Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol 2012;24:398–404.
- Strober W, Murray PJ, Kitani A, Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 2006;6:9–20.
- Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol 2011;4:484–495.
- Conti BJ, Davis BK, Zhang J, CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J Biol Chem 2005;280:18375–18385.
- Zhao Y, Shao F. NLRC5: a NOD-like receptor protein with many faces in immune regulation. Cell Res 2012;22:1099–1101.
- Poeck H, Ruland J. SYK kinase signaling and the NLRP3 inflammasome in antifungal immunity. J Mol Med (Berl) 2010;88:745–752.
- Wen H, Ting JP, O'Neill LA. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat Immunol 2012;13:352–357.
- Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol 2012;20:139–146.
- Satoh T, Kato H, Kumagai Y, LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA 2010;107:1512–1517.
- Kawai T, Takahashi K, Sato S, IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005;6:981–988.
- Takahashi K, Kawai T, Kumar H, Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol 2006;176:4520–4524.
- Dixit E, Boulant S, Zhang Y, Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010;141:668–681.
- Geijtenbeek TB, van Vliet SJ, Engering A, Self- and nonself-recognition by C-type lectins on dendritic cells. Ann Rev Immunol 2004;22:33–54.
- Zelensky AN, Gready JE. The C-type lectin-like domain superfamily. The FEBS J 2005;272:6179–6217.
- Sancho D, Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Ann Rev Immunol 2012;30:491–529.
- Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nature Rev Immunol 2009;9:465–479.
- Huysamen C, Brown GD. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol Lett 2009;290:121–128.
- Tsang E, Giannetti AM, Shaw D, Molecular mechanism of the Syk activation switch. J Biol Chem 2008;283:32650–32659.
- Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harbor Perspectives Biol 2011;3: 10.1101 /cshperspect.a002352.
- Drummond RA, Saijo S, Iwakura Y, The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol 2011;41:276–281.
- Sattler M, Verma S, Pride YB, SHIP1, an SH2 domain containing polyinositol-5-phosphatase, regulates migration through two critical tyrosine residues and forms a novel signaling complex with DOK1 and CRKL. J Biol Chem 2001;276:2451–2458.
- Svajger U, Anderluh M, Jeras M, C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal 2010;22:1397–1405.
- Gringhuis SI, den Dunnen J, Litjens M, Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol 2009;10:1081–1088.
- Gringhuis SI, den Dunnen J, Litjens M, C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 2007;26:605–616.
- Takahara K, Tokieda S, Nagaoka K, C-type lectin SIGNR1 enhances cellular oxidative burst response against C. albicans in cooperation with Dectin-1. Eur J Immunol 2011;41:1435–1444.
- Kerrigan AM, Brown GD. Syk-coupled C-type lectins in immunity. Trends Immunol 32:151–156.
- Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature 2001;413:36–37.
- Ariizumi K, Shen GL, Shikano S, Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem 2000;275:20157–20167.
- Vautier S, Drummond RA, Redelinghuys P, Dectin-1 is not required for controlling Candida albicans colonization of the gastrointestinal tract. Infect Immun 2012;80:4216–4222.
- Hardison SE, Brown GD. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol 2012;13:817–822.
- Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol 2006;6:799–812.
- Gringhuis SI, Wevers BA, Kaptein TM, Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog 2011;7:e1001259.
- Gringhuis SI, den Dunnen J, Litjens M, Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol 2009;10:203–213.
- Underhill DM, Rossnagle E, Lowell CA, Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005;106:2543–2550.
- Fric J, Zelante T, Wong AY, NFAT control of innate immunity. Blood 2012;120:1380–1389.
- Goodridge HS, Underhill DM. Fungal recognition by TLR2 and Dectin-1. Handb Exp Pharmacol 2008:87–109.
- Taylor PR, Brown GD, Herre J, The role of SIGNR1 and the beta-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J Immunol 2004;172:1157–1162.
- Dennehy KM, Ferwerda G, Faro-Trindade I, Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 2008;38: 500–506.
- Mantegazza AR, Barrio MM, Moutel S, CD63 tetraspanin slows down cell migration and translocates to the endosomal-lysosomal-MIICs route after extracellular stimuli in human immature dendritic cells. Blood 2004;104:1183–1190.
- Li X, Utomo A, Cullere X, The beta-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011;10:603–615.
- Sancho D, Mourao-Sa D, Joffre OP, Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest 2008;118:2098–2110.
- Caminschi I, Proietto AI, Ahmet F, The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008;112:3264–3273.
- Poulin LF, Salio M, Griessinger E, Characterization of human DNGR-1 + BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 2010;207:1261–1271.
- Zhang JG, Czabotar PE, Policheni AN, The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 2012;36:646–657.
- Ahrens S, Zelenay S, Sancho D, F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012;36:635–645.
- Geijtenbeek TB. Actin’ as a death signal. Immunity 2012;36:557–559.
- Zelenay S, Keller AM, Whitney PG, The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest 2012;122:1615–1627.
- Huysamen C, Willment JA, Dennehy KM, CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol Chem 2008;283:16693–16701.
- Sancho D, Joffre OP, Keller AM, Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009;458:899–903.
- Colonna M, Samaridis J, Angman L. Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol 2000;30:697–704.
- Suzuki-Inoue K, Fuller GL, Garcia A, A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 2006;107:542–549.
- Hughes CE, Sinha U, Pandey A, Critical role for an acidic amino acid region in platelet signalling by the hemITAM (hemi-Immunoreceptor Tyrosine-based Activation Motif) containing receptor CLEC-2 (C-type Lectin receptor-2). J Biol Chem 2012;288(7):5127–5135.
- Fuller GL, Williams JA, Tomlinson MG, The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem 2007;282:12397–12409.
- Watson AA, Brown J, Harlos K, The crystal structure and mutational binding analysis of the extracellular domain of the platelet-activating receptor CLEC-2. J Biol Chem 2007;282:3165–3172.
- Kato Y, Kaneko MK, Kunita A, Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci 2008;99:54–61.
- Suzuki-Inoue K, Kato Y, Inoue O, Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem 2007;282:25993–26001.
- Pollitt AY, Grygielska B, Leblond B, Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood 2010;115:2938–2946.
- Kerrigan AM, Dennehy KM, Mourao-Sa D, CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J Immunol (Baltimore, MD: 1950) 2009;182:4150–4157.
- Hughes CE, Pollitt AY, Mori J, CLEC-2 activates Syk through dimerization. Blood 115:2947–2955.
- Christou CM, Pearce AC, Watson AA, Renal cells activate the platelet receptor CLEC-2 through podoplanin. Biochem J 2008;411:133–140.
- Pleines I, Elvers M, Strehl A, Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch 2009;457:1173–1185.
- Mourao-Sa D, Robinson MJ, Zelenay S, CLEC-2 signaling via Syk in myeloid cells can regulate inflammatory responses. Eur J Immunol 2011;41:3040–3053.
- Redelinghuys P, Brown GD. Inhibitory C-type lectin receptors in myeloid cells. Immunol Lett 2011;136:1–12.
- Marshall AS, Willment JA, Lin HH, Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J Biol Chem 2004;279:14792–14802.
- van Rhenen A, van Dongen GA, Kelder A, The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007;110:2659–2666.
- Zhao X, Singh S, Pardoux C, Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2009;95:71–78.
- Chen CH, Floyd H, Olson NE, Dendritic-cell-associated C-type lectin 2 (DCAL-2) alters dendritic-cell maturation and cytokine production. Blood 2006;107:1459–1467.
- Han Y, Zhang M, Li N, KLRL1, a novel killer cell lectin like receptor, inhibits natural killer cell cytotoxicity. Blood 2004;104:2858–2866.
- Sattler S, Ghadially H, Reiche D, Evolutionary development and expression pattern of the myeloid lectin-like receptor gene family encoded within the NK gene complex. Scand J Immunol 2010;72:309–318.
- Hoffmann SC, Schellack C, Textor S, Identification of CLEC12B, an inhibitory receptor on myeloid cells. J Biol Chem 2007;282:22370–22375.
- Sobanov Y, Bernreiter A, Derdak S, A novel cluster of lectin-like receptor genes expressed in monocytic, dendritic and endothelial cells maps close to the NK receptor genes in the human NK gene complex. Eur J Immunol 2001;31:3493–3503.
- Sattler S, Reiche D, Sturtzel C, The human C-type lectin-like receptor CLEC-1 is upregulated by TGF-beta and primarily localized in the endoplasmic membrane compartment. Scand J Immunol 2012;75:282–292.
- Thebault P, Lhermite N, Tilly G, The C-type lectin-like receptor CLEC-1, expressed by myeloid cells and endothelial cells, is up-regulated by immunoregulatory mediators and moderates T cell activation. J Immunol 2009;183:3099–3108.
- Li Y, Duan Z, Gao D, The new role of LOX-1 in hypertension induced neuronal apoptosis. Biochem Biophys Res Commun 425:735–740.
- Shimaoka T, Kume N, Minami M, LOX-1 supports adhesion of gram-positive and gram-negative bacteria. J Immunol 2001;166:5108–5114.
- Mitra S, Goyal T, Mehta JL. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther 2011;25:419–429.
- Hayashida K, Kume N, Murase T, Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome - A novel marker for early diagnosis. Circulation 2005;112:812–818.
- OBrien PJ, Dameron GW, Beck ML, Cardiac troponin T is a sensitive, specific biomarker of cardiac injury in laboratory animals. Lab Animal Sci 1997;47:486–495.
- Pant S, Deshmukh A, Mehta JL. The LOX-1 scavenger receptor and its implication in coronary artery disease. Clin Lipidol 2012;7:271–273.
- Zhu H, Lee C, Zhang D, Surface-associated GroEL facilitates the adhesion of Escherichia coli to macrophages through lectin-like oxidized low-density lipoprotein receptor-1. Microbes Infect 2012. http://dx.doi.org/10.1016/j.micinf.2012.10.001.
- Dunn S, Vohra RS, Murphy JE, The lectin-like oxidized low-density-lipoprotein receptor: a pro-inflammatory factor in vascular disease. Biochem J 2008;409:349–355.
- Li D, Mehta JL. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 2000;101:2889–2895.
- Li D, Mehta JL. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circ Res 2009;104:566–568.