Abstract
Background: Many efforts have been made to investigate the role played by cytokines in the development of hypertension. But few reports exist on the association between cytokines and hypertensive renal damage. This study was to observe the changes of the serum levels of cytokines, tumor necrosis factor alpha (TNF-α), and interleukin 6 (IL-6) in patients with hypertensive renal damage, whereby to study the correlation of TNF-α and IL-6 with the hypertensive renal damage. Methods: According to their urinary albumin excretion rate (UAER), 102 patients with essential hypertension were divided into three groups: normoalbuminuria hypertensive group (n = 37), microalbuminuria hypertensive group (n = 36), and proteinuric hypertensive group (n = 29). Serum TNF-α and IL-6 of all subjects were measured with radioimmune assay. Thirty age- and gender-matched normotensive persons were selected as normotensive control group. Results: Serum levels of TNF-α and IL-6 were significantly higher in patients with essential hypertension than those in normotensive control group (p < 0.5). Serum levels of TNF-α and IL-6 increased in proportion to UAER. The statistical significance was present among groups (p < 0.05). Both TNF-α and IL-6 were found to have a positive correlation with UAER (r = 0.79, p < 0.01; r = 0.75, p < 0.01), but not with the levels of blood pressure (BP). Conclusions: TNF-α and IL-6 are remarkably increased in hypertensive patients and may play an important role in the pathogenesis and the development of hypertensive renal damage.
INTRODUCTION
Heart, brain, and kidney are the most important target organs of hypertension, and one of the stages of the injury by hypertension of them are hypertensive heart disease, stroke, and hypertensive renal damage, respectively. Previous studiesCitation1–4 have shown that in patients with hypertension there was an increased production of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), which might play an important role in the initiation and progression of hypertension by a variety of mechanisms such as giving rise to contraction of vessels, leading to thickening of vascular wall, and proliferation of endothelial cells and smooth muscle cells. Despite such advances as mentioned above, to the best of our knowledge, few reports exist on the role played by cytokines in the initiation and progression of hypertensive renal damage. The aim of this study was to investigate the role played by cytokines in the development and progression of hypertensive renal damage through measurement of serum levels of TNF-α, IL-6, and urinary protein in patients with essential hypertension.
SUBJECTS AND METHODS
Subjects
In light of the diagnostic standards of essential hypertension issued by World Health Organization and International Society of Hypertension in 1999, 102 patients with essential hypertension who were admitted from July 2008 to December 2008 were enrolled, 60 male and 42 female, and the mean age was 56.7 ± 11.2 years. The levels of blood pressure (BP) were classified as class 1 or class 2. All of them agreed to take part in this study and written informed consent was provided.
Based on their urinary albumin excretion rate (UAER), the subjects were divided into three groups: normoalbuminuria hypertensive group (UAER < 20 μg/min, 37 cases, 22 male and 15 female, mean age 51.4 ± 13.4 years), microalbuminuria hypertensive group (20 μg/min < UAER < 200 μg/min, 36 cases, 19 male and 17 female, mean age 54.8 ± 10.6 years), and proteinuric hypertensive group (UAER > 200 μg/min, 29 cases, 19 man and 10 female, mean age 53.2 ± 13.2 years).
Patients with secondary hypertension, coronary artery disease, diabetes mellitus, severe cardio- and cerebrovascular disease, liver dysfunction, renal dysfunction, cardiac valvular disease, heart failure, autoimmune disease, tumor, infection, use of immunomodulator, endocrine disease, and metabolic disease were excluded. Smoking and drinking in subjects were suspended for at least 3 months before the study was undertaken.
Simultaneously, 30 healthy persons with normal BP were selected as normotensive control group, 17 males and 13 females, and the mean age was 50.5 ± 11.1 years.
Sampling and measurement
Five milliliter of venous blood for each subject was collected after half an hour of quiet lying in the morning after 8 hours of fasting and was then centrifuged at 3000 rpm (2264 × g) for 10 min at 4°C, and stored at −70°C for measurement of blood parameters.
Both systolic blood pressure (SBP) and diastolic blood pressure (DBP) were measured after at least 5 min of rest and the two values averaged. Hypertension was defined as an SBP of 140 mmHg or greater and/or a DBP of 90 mmHg or greater that occurred as the average of the second and third measures on the initial clinic visit. Participants were also identified as hypertensive if they self-reported the presence of hypertension and had or being used any antihypertensive medication. Body mass index (BMI) was calculated as body weight divided by the square of body height.
The measurement of UAER was by radioimmunoassay: the 24-hour urinary sample was collected twice with an interval of more than 1 week. After the total volume was calculated, 5 mL of urinary sample from each subject was taken and centrifuged. Then supernatant was separated and stored at −20°C for measurement of urinary protein with radioimmunoassay. The UAER was calculated as the urinary protein divided by the total volume of 24-hour urinary sample.
Both TNF-α and IL-6 were detected by radioimmunoassay in accordance with the instructional manual of the kits. The sensitivity of the kit of TNF-α was <0.3 ng/mL and IL-6 <50 pg/mL. Measurement of blood urea nitrogen (BUN) and creatinine (Cr) was performed using Berthelot enzymatic-colorimetric assay and enzymatic assay, respectively. Plasma glucose levels were determined using the glucose oxidase method. Serum total cholesterol and serum triglyceride were measured with CHOD-PAP and GPO-POD, respectively.
Statistics
Statistical package for social sciences (SPSS, version 11.5) was used to process the data. Continuous variables are expressed as the mean ± SEM and categorical variables are described in terms of frequencies and percentages. The data were analyzed with t-test when means between two groups were compared, and with one-way analysis of variance (ANOVA) plus Turkey's post hoc multiple comparison test when mean values across multiple groups were compared. Any association between inflammatory marker and UAER was tested by calculating bivariate Pearson's correlation coefficients. A p-value less than 0.05 denoted the presence of a statistically significant difference.
RESULTS
Baseline data
No statistically significant difference was found in terms of gender, BMI, blood glucose, lipid profile, BUN, and Cr between four groups (p > 0.05) ( and ).
TABLE 1. Comparison of age, gender, blood pressure, and BMI among groups
TABLE 2. Comparison of glucose, cholesterol, triglyceride, urea nitrogen, and creatinine among groups
UAER, TNF-α, and IL-6 between groups
There was no significant difference in UAER between normotensive group and normoalbuminuria hypertensive group (p > 0.05) (). The UAER in microalbuminuria hypertensive group and proteinuric hypertensive group was notably higher than that in normotensive group and normoalbuminuria hypertensive group (p < 0.05), and, of course, the UAER in microalbuminuria hypertensive group was lower than that in proteinuric hypertensive group (p < 0.05). The levels of TNF-α and IL-6 in hypertensive group were higher than those in normotensive control group (p < 0.05) and increased with the increasing UAER (p < 0.05) ().
TABLE 3. Comparison of UAER, TNF-α and IL-6 among groups
Correlation of TNF-α and IL-6 with UAER
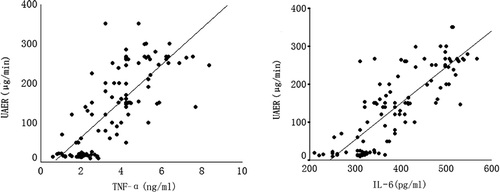
Both TNF-α and IL-6 have a positive correlation with UAER (r = 0.79, p < 0.01; r = 0.75, p < 0.01), but no correlation with the level of BP ().
FIGURE 1. Correlation of TNF-α and IL-6 with UAER. Abbreviations: UAER, urinary albumin excretion rate; TNF-α, tumor necrosis factor alpha; IL-6, interleukin 6.
DISCUSSION
Our study demonstrated that under the condition of similar age, BMI, glucose, lipids, urea nitrogen, and creatinine, the patients with hypertension had a higher level of TNF-α and IL-6 than the normotensives suggesting that TNF-α and IL-6 may play a role in the development of hypertension by the possible mechanisms such as cellular immune dysfunction and inflammatory reaction.Citation5,Citation6
Some reportsCitation3,Citation4,Citation7 have shown a significant association between inflammatory markers and elevated BP, consistent with our findings. Kim et al.Citation8 reported that TNF-α level correlated with the BP index and IL-6 level correlated with the BP variability index, implying that inflammation may be a mediator for the link between BP variability and target organ damage. In two-kidney, one-clip (2K1C) hypertension rats, plasma cytokines such as IL-6 and TNF-α were higher than in control.Citation9 TNF-α has been reported to activate a variety of vasoconstrictors such as endothelin and platelet-derived growth factor (PDGF) from cultured endothelial cellsCitation10–13 and to impair the function of endothelium-dependent vasodilatation. A modest elevation of TNF-α has been shown to destabilize the mRNA of endothelial nitric oxide synthase (eNOS). IL-6 can also increase the production of C-reactive protein in liver of which in its turn decreases the production of endogenous nitric oxide and can induce the synthesis of PDGF that have the function of leading to the contraction of vascular smooth muscle cells and the proliferation of fibroblasts.Citation14
In addition, PDGF increased by TNF-α and IL-6 promotes the reactive sensitivity of artery wall to low-density lipoprotein, pertains to the contraction of vascular wall and to escalation of BP, and increases the expression of adhesion molecule of vascular endothelial cells (VEC), which may in turn harm the VEC, both directly and indirectly, and thus lead to the decrease of the synthesis and secretion of vasodilators and the increase of vasoconstrictors characteristic of VEC dysfunction.Citation15 Naya et al.Citation16 also showed that IL-6 and TNF-α were correlated with coronary vascular resistance (CVR) during a cold pressor test (CPT) in hypertensive patients and were significant independent predictors of CVR during CPT indicating elevated plasma IL-6 and TNF-α levels were independent predictors of coronary endothelial dysfunction in hypertensive patients.
Furthermore, the increased angiotensin II in hypertension may increase transcription and synthesis of proinflammatory cytokines such as TNF-α and IL-6, which in turn activate the synthesis of angiotensinogen, whereby lead to the overexpression at downstream of angiotensin that finally contracts the vascular and facilitates hyperplasia.Citation17–21
IL-6 and TNF-α also significantly increase the inward flow of Ca2+ by increasing Ca2+-dependent K+ outflow and promoting the permeability of cell membrane to Ca2+. The increased inward flow of Ca2+ leads to the contraction of vascular smooth muscular cells (VSMC) which escalates BP.Citation22 Circulating TNF-α, directly or indirectly, stimulates the abnormal expression through endothelin from endothelial cells of proto-oncogene such as c-sis, c-myc, and c-fos that may promote the synthesis of DNA and the proliferation of VSMC, thus leading to the thickening of vascular wall, narrowing of lumen, increasing of peripheral resistance, and finally promoting of BP. The study by Azra Mahmud et al.Citation23 also demonstrated that TNF-α and IL-6 were related to arterial stiffness. The net effect of function mentioned above is the increase of peripheral vascular resistance characterizing hypertension.
Our study has also shown that the serum levels of TNF-α and IL-6 in proteinuric hypertensive group were significantly higher than that in both microalbuminuria hypertensive group and proteinuric hypertensive group; furthermore, the serum level of TNF-α and IL-6 in microalbuminuria hypertensive group was obviously higher than that in normoalbuminuria hypertensive group. Linear correlation analysis indicated that the serum level of TNF-α and IL-6 had a positive correlation with the UAER, but not with BP, showing that TNF-α and IL-6 may take part in the progression of hypertensive damage and may be independent risk factors and prognostic predictors of hypertensive renal damage.Citation24,Citation25 Both increased IL-6 and TNF-α can upgrade the immune reaction which may pertain to the overproduction of immune compounds that can directly increase the glomerular permeability and activate the complements. These activated complements can lead to the production of chemoattractants such as C3a and C5a that attract inflammatory cells and make them release proinflammatory factors, proteinase, reactive oxygen species, and so on, and, by the function of these products, bring forth the renal damage.
IL-6 can induce the infiltration in the renal interstitium of lymphocyte that played a key role in the immune-mediated renal disease.Citation26 TNF-α can contribute to influence the coagulation system and fibrinolytic system by a variety of mechanisms including inducing the platelet to aggregate on the endothelial to form thrombi, inducing the mesangial cells to produce active substance similar to tissue factor, and increasing the activity of preclotting substances.
Certain studies have shown that TNF-α can stimulate the mesangial cells to produce oxygen-free radicals and whereby to increase the production of lipid peroxide metabolites that then lead to the injury of cell membrane.Citation27
Furthermore, IL-6 and TNF-α can also induce the contraction and proliferation of mesangial cells,Citation28–30 deposit of matrix, acceleration of glomerulosclerosis, and stimulate the proliferation of the fibroblast, whereby to produce collagen fibers which can finally lead to renal fibrosis.
IL-6 can also stimulate synthesis and excretion of colloid enzyme and other extracellular enzyme produced by the matrix cells, which can lead to the degradation of negatively charged glycoprotein that is located at basement membrane and thus lead to the leakage of negatively charged protein from plasma and whereby to result in proteinuria.Citation31
Acknowledgment
This study was partly supported by the Ningxia Medical University General Projects Research Fund (No. 200900).
REFERENCES
- Cymerys M, Chyrek R, Bogdanski P, Lacki J, Pupek-Musialik D. Evaluation of acute phase proteins in hypertensive and obese patients. Pol Merkuriusz Lek. 2003;15:352–355.
- Bogdanski P, Kujawska-Luczak M, Lacki J, Pupek-Musialik D. Evaluation of selected interleukins, tumor necrosis factor, insulin and leptin in obese patient with hypertension. Pol Merkuriusz Lek. 2003;15:347–351.
- Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154.
- Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001; 38:399–403.
- Cottone S, Vadalà A, Vella MC, Mulé G, Contorno A, Cerasola G. Comparison of tumor necrosis factor and endothelin-1 between essential and renal hypertension patients. J Hum Hypertens. 1998;12:351–354.
- Dzielak DJ. The immune system and hypertension. Hypertension. 1992;19(Suppl. 1):I36–I44.
- Sardo MA, Mandraffino G, Campo S, Biglycan expression in hypertensive subjects with normal or increased carotid intima-media wall thickness. Clin Chim Acta. 2009; 406(1–2):89–93.
- Kim KI, Lee JH, Chang HJ, Association between blood pressure variability and inflammatory marker in hypertensive patients. Circ J. 2008;72:293–298.
- Bivol LM, Berge RK, Iversen BM. Tetradecylthioacetic acid prevents the inflammatory response in two-kidney, one-clip hypertension. Am J Physiol Regul Integr Comp Physiol. 2008; 294:R438–R447.
- Conrad KP, Benyo DF. Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol. 1997;37:240–249.
- Marsden PA, Brenner BM. Transcriptional regulation of the endothelin-1 gene by TNF-α. Am J Physiol. 1992;262: C854–C861.
- Hajjar KA, Hajjar DP, Silverstein RL, Nachman RL. Tumor necrosis factor-mediated release of platelet-derived growth factor from cultured endothelia cells. J Exp Med. 1987;166: 235–245.
- Van Hinsbergh VW, Kooistra T, van den Berg EA, Princen HM, Fiers W, Emeis JJ. Tumor necrosis factor increases the production of plasminogen activator inhibitor in human endothelial cells in vitro and rats in vivo. Blood. 1988;72:1467–1473.
- Ikeda U, Ikeda M, Oohara T, Interleukin 6 stimulates growth of vascular smooth muscle cells in a PDGF-dependent manner. Am J Physiol. 1991;260:H1713–H1717.
- Yoshizumi M, Perrella MA, Burnett JC Jr, Lee ME. Tumor necrosis factor down regulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73:205–209.
- Naya M, Tsukamoto T, Morita K, Plasma interleukin-6 and tumor necrosis factor-alpha can predict coronary endothelial dysfunction in hypertensive patients. Hypertens Res. 2007;30:541–548.
- Han Y, Runge MS, Brasier AR. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor kappa B transcription factors. Circ Res. 1999;84:695–703.
- Skurk T, van Harmelen V, Hauner H. Angiotensin II stimulates the release of interleukin-6 and interleukin-8 from cultured human adipocytes by activation of nuclear factor kappa B. Arterioscler Thromb Vasc Biol. 2004;24:1199–1203.
- Kranzhöfer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kübler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1623–1629.
- Brasier AR, Li J, Wimbish KA. Tumor necrosis factor activates angiotensinogen gene expression by Rel A transactivator. Hypertension. 1996;27:1009–1017.
- Takano M, Itoh N, Yayama K, Yamano M, Ohtani R, Okamoto H. Interleukin-6 as a mediator responsible for inflammation-induced increase in plasma angiotensinogen. Biochem Pharmacol. 1993;45:201–206.
- Buemi M, Marino D, Floccari F, Effect of interleukin-8 and ICAM-1 on calcium-dependent outflow of K+ in erythrocytes from subjects with essential hypertension. Curr Med Res Opin. 2004;20:19–24.
- Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46: 1118–1122.
- Simmons EM, Himmelfarb J, Sezer MT, Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int. 2004;65:1357–1365.
- Pecoits-Filho R, Bárány P, Lindholm B, Heimbürger O, Stenvinkel P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol Dial Transplant. 2002;17:1684–1688.
- Coletta I, Soldo L, Polentarutti N, Selective induction of MCP-1 in human mesangial cells by the IL-6/sIL-6R complex. Exp Nephrol. 2000;8:37–43.
- Nanami M, Ookawara T, Otaki Y, Tumor necrosis factor alpha induced iron sequestration and oxidative stress in human endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:2495–2501.
- Sterzel RB, Schulze-Lohoff E, Marx M. Cytokines and mesangial cells. Kidney Int Suppl. 1993;39:26–31.
- Ruef C, Budde K, Lacy J, Interleukin 6 is an autocrine growth factor for mesangial cells. Kidney Int. 1990; 38:249–257.
- Horii Y, Muraguchi A, Iwano M, Involvement of IL-6 in mesangial proliferative glomerulonephritis. J Immunol. 1989; 143:3949–3955.
- Ritz E, Stefanski A. Diabetic nephropathy in type2 diabetes. Am J Kidney Dis. 1996;27:167–194.