Abstract
Herein, we report a case of acute kidney injury (AKI) due to diarrhea-induced acute tubular necrosis (ATN) in a patient with nephrotic syndrome secondary to biopsy-proven collapsing focal and segmental glomerulosclerosis (FSGS). The clinical picture mimicked rapidly progressive glomerulonephritis (RPGN) and motivated pulse therapy with methylprednisolone and cyclophosphamide. The case presentation is followed by a brief overview of the epidemiology of AKI in nephrotic syndrome as well as a discussion of its risk factors and potential mechanisms involved.
INTRODUCTION
Clinical–pathologic correlation allow nephrologists to group the immune-mediated glomerular diseases into two main categories: (1) those where the immune deposits predominantly affect the sub-endothelial space, leading to an inflammatory (or nephritic) syndrome characterized by hypertension, hematuria, proteinuria, edema, and renal dysfunction; and (2) those where the immune deposits predominantly affect the sub-epithelial space, resulting in a severely proteinuric (or nephrotic) syndrome typified by marked hypoalbuminemia, edema, and hyperlipidemia. Although proteinuria and edema are shared by these two syndromes, they both tend to be more intense in nephrotic than in nephritic cases. It is, therefore, possible to predict the type of glomerular disease and sometimes even “guess” the exact histology based on the clinical presentation; academic nephrologists have been doing this for many years at clinical–pathologic correlation conferences. Practicing nephrologists also engage on the same exercise before prescribing empiric treatment while awaiting renal biopsy results.
Yet, some patients may present with clinical manifestations that cannot be readily grouped into one of these two polar spectra. Sometimes the very nature of the glomerular disease leads to a mixed nephritic/nephrotic pattern, as is the case with membranoproliferative glomerulonephritis or lupus membranous nephritis with proliferation. In other circumstances, a purely nephrotic lesion is complicated by acute kidney injury (AKI) due to drug toxicity, effective arterial volume depletion, or acute tubular necrosis (ATN).Citation1 These cases often present themselves as diagnostic challenges and may lead to incorrect empiric treatment. Herein, we report a case of AKI secondary to ATN in a patient with collapsing focal and segmental glomerulosclerosis (FSGS) mimicking rapidly progressive glomerulonephritis (RPGN).
MATERIALS AND METHODS
Case report and review of the literature.
RESULTS
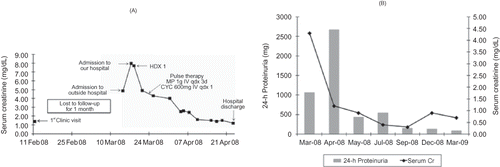
A 49-year-old Brazilian female was transferred from a small community hospital to a tertiary care facility for emergent hemodialysis. Transfer papers indicated that she had presented to the outside hospital with a 1-week history of fever, abdominal pain, diarrhea, and oliguria. In addition, she admitted to a 4-month history of lower extremity edema progressing to anasarca, for which she was evaluated 1 month prior to admission. At that time, she was found to have a normal blood pressure, facial and lower extremity edema, BUN 32 mg/dL, serum creatinine 1.38 mg/dL, and urinalysis with 3+ protein and a benign sediment. She denied any further investigation or treatment for this condition. Her medical history was otherwise unremarkable and she denied taking any medications. Upon admission to the outside hospital, she was afebrile and normotensive; orthostatic changes were not reported. Physical examination revealed anasarca and bilateral pleural effusions; there was no jaundice. Laboratory studies showed urinalysis with 3+ protein, 6 white blood cells, and too numerous to count red blood cells per high power field but no red cell casts; there were no eosinophils in the urine; BUN 43 mg/dL, serum creatinine 4.86 mg/dL, hematocrit 28%, white blood cell count 15,700 without bandemia and platelet count 447,000; a peripheral blood smear was negative for schistocytes; stool studies or blood cultures were not performed. Infectious diarrhea and AKI were suspected and she was started on intravenous antibiotics and gentle volume expansion with crystalloids. Three days later, diarrhea had resolved but BUN and serum creatinine had risen to 70 and 7.9 mg/dL, respectively, and she was transferred to a tertiary care facility for emergent hemodialysis. Further investigation included normal serum complements, negative viral and auto-antibody panels, 24-hour urine protein 1059 mg, serum albumin < 1.0 g/dL, total cholesterol 306 mg/dL, and triglycerides 208 mg/dL. Renal ultrasound revealed normal-sized kidneys. Given the history of proteinuria, hematuria, and rapid loss of renal function, a diagnosis of RPGN was entertained. Treatment was started with pulse therapy with methylprednisolone (1 g IV per day for 3 days) and cyclophosphamide (600 mg IV, single dose) followed by prednisone 1 mg/kg/day. A percutaneous renal biopsy was performed without complications and revealed collapsing FSGS with a component of ATN (). She experienced a quick recovery of renal function and was able to discontinue dialysis after a single session (). She was discharged 4 weeks later with a serum creatinine of 1.2 mg/dL and 24-hour urine protein of 2670 mg. One month later, serum creatinine was 0.9 mg/dL and 24-hour urine protein was down to 430 mg. Five months after discharge, 24-hour urine protein was down to 147 mg and a steroid taper was begun. One year later, she remains in complete remission with a serum creatinine of 0.7 mg/dL and 24-hour urine protein of 89 mg ().
FIGURE 1. Kidney histology showing focal segmental glomerular sclerosis. (A–D) Most glomeruli and tubules are normal (A). Some glomeruli show segmental collapse and overlying podocyte hypertrophy and hyperplasia (B and C). Patches of injured tubules showing flattening and loss of tubule brush border; cellular debris are seen in the lumen. The interstitium is edematous with a few leukocytes (D).
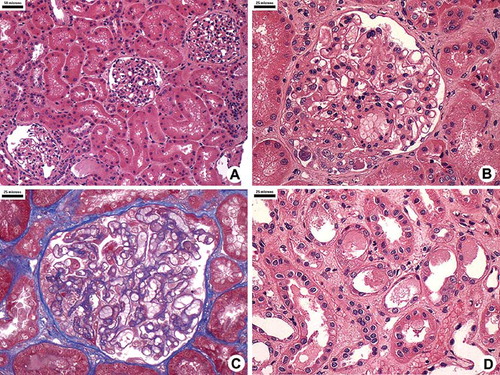
FIGURE 2. (A) Evolution of serum creatinine levels from initial clinic visit till hospital discharge. HD, hemodialysis; MP, methylprednisolone; CYC, cyclophosphamide. (B) One-year follow-up of serum creatinine levels and 24-hour urine protein excretion.
DISCUSSION
Although the loss of renal function coupled with edema, proteinuria, and active urinary sediment resembled RPGN, several other features were not entirely consistent with this diagnosis. First, the loss of renal function was too brisk and, as shown in , the slope of the rise in serum creatinine levels was more compatible with an ATN than with a glomerulonephritis.Citation2 Second, the severity of the edema, hypoalbuminemia, and the presence of dyslipidemia suggested a nephrotic rather than a nephritic picture. Third, the finding of a normal blood pressure argued against RPGN. Finally, the 4-month history of edema preceding the current presentation pointed toward a more insidious glomerular disease. The patient received pulse therapy with steroids and cyclophosphamide but other diagnoses were then entertained. Given the history of diarrhea, we first considered hemolytic uremic syndrome (HUS). HUS typically follows infection with a Shiga toxin-producing E. coli, usually of the O157:H7 serotype, and presents with microangiopathic hemolytic anemia, thrombocytopenia, and AKI.Citation3 Although we did not have LDH levels or a reticulocyte count, HUS was excluded given the normal platelet count and absence of schistocytes in the peripheral blood smear. Allergic interstitial nephritis due to the antibiotics taken for diarrhea was considered unlikely given the absence of eosinophiluria and the presence of the nephrotic syndrome. In fact, several features pointed toward a nephrotic lesion, such as minimal change disease (MCD), FSGS, or membranous nephropathy. On the other hand, the 24-hour proteinuria was less than 3.5 g and the presence of AKI and active urine sediment suggested a more inflammatory renal lesion. One consideration would be membranous nephropathy with ANCA-associated necrotizing and crescentic glomerulonephritis, a rare dual glomerulopathy that can present with heavy proteinuria, AKI, and active urine sediment;Citation4 since ANCA positivity is seen in almost all patients with this entity, the negative ANCA in our case argued against this possibility. Another type of “mixed lesion,” such as lupus membranous nephropathy with proliferation, was ruled out given the negative auto-antibodies and lack of other criteria for systemic lupus erythematosus. We were left considering a “purely nephrotic lesion” complicated by AKI. The renal biopsy showed collapsing FSGS with superimposed ATN and helped to reconcile all of the findings.
We encountered very few reports focusing on AKI complicating the course of FSGS.Citation5–7 AKI has been described as a rare complication of the nephrotic syndrome in adult patients with MCD. In 1990, Jennette and Falk performed a case–control study comparing 21 adults with MCD and a serum creatinine greater than 2.0 mg/dL with 50 adults with MCD and a serum creatinine less than 1.5 mg/dL. They found that patients with AKI were older, and had higher systolic blood pressure, more proteinuria, and more arteriosclerosis in the renal biopsy specimen; ATN was observed in 71% of the patients with AKI and 0% of those without.Citation8 Two years later, Smith and Hayslett reviewed 79 cases of AKI associated with MCD reported in the literature and arrived at similar conclusions; they stated that AKI in patients with MCD “may result from an interaction between acute ischemic tissue injury and preexisting intrinsic renal abnormalities.”Citation9 A more recent study on adult MCD by Waldman et al. compared the features of 24 patients with AKI with those of 75 without. Likewise, they found that patients with AKI tended to be older and hypertensive, with lower serum albumin and more proteinuria than those without AKI; interestingly, AKI patients were more likely to have had resistance to steroids and to have FSGS on a repeat renal biopsy.Citation10
In a review of the pathophysiology of AKI in patients with idiopathic nephrotic syndrome, Koomans suggested the following as potential mechanisms: decreased perfusion pressure, decreased filtration coefficient, high intra-tubular pressure, ATN, interstitial nephritis, and interstitial edema.Citation1 Our patient had a very low albumin and anasarca; in this setting, the additional reduction in effective circulating arterial volume induced by the diarrhea probably led to ischemic ATN. The ATN findings in our patient's biopsy were patchy and mild despite profound renal dysfunction; this physiologic and pathologic dissociation has been previously described.Citation11
It could be argued that our patient's 24-hour proteinuria < 3.5 g and presence of hematuria are still left unexplained. We posit that the first 24-hour urine collection was performed during oliguric AKI (serum creatinine of 4.3 mg/dL), which may have impaired protein excretion; nevertheless, a laboratory error cannot be excluded. A second 24-hour urine collection performed 1 month later, while the serum creatinine was 1.2 mg/dL, also revealed sub-nephrotic proteinuria but this can be simply attributed to a response to therapy. In our opinion, the presentation with anasarca and serum albumin level < 1.0 g/dL leaves no doubt as to the severity of our patient's proteinuria. Although FSGS typically presents with “pure” nephrotic syndrome, the presence of microscopic hematuria is not that uncommon. Indeed, in one study, some degree of hematuria was detected in the majority of FSGS patients.Citation12 Other authors have shown that 12–19% of patients with FSGS may actually present with a nephritic picture.Citation13–15 At least in children with FSGS, the presence of hematuria has been linked to a higher risk of progression to chronic kidney disease.Citation16
This report is a reminder of how AKI may complicate the course of nephrotic syndrome and create a diagnostic challenge. In our case, the combined presence of AKI and hematuria in a patient with collapsing FSGS mimicked an RPGN and prompted aggressive therapy with steroids and cyclophosphamide. Nevertheless, careful interpretation of the history and laboratory findings allows anticipation of the biopsy findings even in complex cases, with blending of pathologies.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Koomans HA. Pathophysiology of acute renal failure in idiopathic nephrotic syndrome. Nephrol Dial Transplant. 2001; 16(2):221–224.
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334(22):1448–1460.
- Tarr PI. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: Distinct mechanisms of pathogenesis. Kidney Int Suppl. 2009 February(112):S29–S32.
- Nasr SH, Said SM, Valeri AM, Membranous glomerulonephritis with ANCA-associated necrotizing and crescentic glomerulonephritis. Clin J Am Soc Nephrol. 2009;4(2): 299–308.
- Futrakul N, Siriviriyakul P, Deekajorndej T, Futrakul P. Hemodynamic maladjustment and disease progression in nephrosis with FSGS. Ren Fail. 2004;26(3):231–236.
- Tanaka H, Tateyama T, Waga S. Acute renal failure at the onset of idiopathic nephrotic syndrome in two children. Clin Exp Nephrol. 2001;5:47–49.
- Polaina Rusillo M, Borrego Utiel FJ, Ruiz Ávila I, Perez Bañasco V. Acute renal failure in a case of nephrotic syndrome secondary to focal and segmental glomerulosclerosis. Nefrologia 2008;28(1):106–107.
- Jennette JC, Falk RJ. Adult minimal change glomerulopathy with acute renal failure. Am J Kidney Dis. 1990;16(5):432–437.
- Smith JD, Hayslett JP. Reversible renal failure in the nephrotic syndrome. Am J Kidney Dis. 1992;19(3):201–213.
- Waldman M, Crew RJ, Valeri A, Adult minimal-change disease: Clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol. 2007;2(3):445–453.
- Rosen S, Stillman IE. Acute tubular necrosis is a syndrome of physiologic and pathologic dissociation. J Am Soc Nephrol. 2008;19(5):871–875.
- Mitwalli AH, Al WJ, bu-Aisha H, Prevalence of glomerular diseases: King khalid university hospital, saudi arabia. Saudi J Kidney Dis Transpl. 2000;11(3):442–448.
- Covic A, Schiller A, Volovat C, Epidemiology of renal disease in Romania: A 10 year review of two regional renal biopsy databases. Nephrol Dial Transpl. 2006;21(2):419–424.
- Rychlik I, Jancova E, Tesar V, The Czech registry of renal biopsies. Occurrence of renal diseases in the years 1994–2000. Nephrol Dial Transpl. 2004;19(12):3040–3049.
- Naini AE, Harandi AA, Ossareh S, Ghods A, Bastani B. Prevalence and Clinical Findings of Biopsy-Proven Glomerulonephritidis in Iran. Saudi J Kidney Dis Transpl. 2007;18(4): 556–564.
- Abrantes MM, Cardoso LS, Lima EM, Predictive factors of chronic kidney disease in primary focal segmental glomerulosclerosis. Pediatr Nephrol. 2006;21(7):1003–1012.