Abstract
Primary hyperoxaluria (PH) is a rare autosomal recessive disease caused by the functional defect of alanine-glyoxylate aminotransferase (AGT) enzyme in the liver and it is characterized by the deposition of diffuse calcium oxalate crystals. A 38-year-old male patient presented with history of recurrent nephrolithiasis and has received chronic hemodialysis treatment for 2 years. Cadaveric renal transplantation was applied to the case. The patient was reoperated on postoperative day 13 because of the collection surrounding the urethra. During this operation, kidney biopsy was made due to late decrease in creatinine levels. Deposition of diffuse oxalate crystal was detected in allograft kidney biopsy, whereas in the 0-hour biopsy there were no oxalate crystals. Oxalate level was found to be high in a 24-hour urine specimen (118 mg/L, normal level: 7–44 mg/L). The patient was identified with primary hyperoxaluria and followed up in terms of systemic oxalate deposition as well as allograft kidney. In the kidney biopsy taken after 18 months, we detected that oxalate crystals almost entirely disappeared. In our case, bilateral preretinal, intraretinal, and intravascular diffuse oxalate crystals were detected, and argon laser photocoagulation treatments were needed for choroidal and retinal neovascularization. Repeated ophthalmic examinations showed the regressive nature of oxalate depositions. In the 18th month, fundus examination and fluorescein angiography revealed that oxalate crystals were significantly regressed. To increase the quality of life and slow down the systemic effects of oxalosis, kidney-only transplantation is beneficial.
INTRODUCTION
Primary hyperoxaluria (PH) is a rare autosomal recessive disease caused by the functional defect of alanine-glyoxylate aminotransferase (AGT) enzyme in the liver and it is characterized by the deposition of diffuse calcium oxalate crystals.Citation1 Progressive calcium oxalate urolithiasis and nephrolithiasis result in renal failure in the course of time leading to oxalate accumulation in soft tissues and bones. Calcium oxalate accumulation and systemic oxalosis are detected in kidneys, myocardium, skeletal and retinal tissues as well as in lymph nodes, coronary artery, and peripheral blood muscles. Bone marrow involvement, digital infarcts, livedo reticularis, large vessel occlusions, retinal and choroidal involvement, cardiomyopathy, heart block, arrhythmias, mononeuritis multiplex, synovitis, and osteosclerosis are among the clinical findings.Citation2
In this article, we will present a patient in whom the diagnosis was made as a result of the detection of oxalate crystals in an allograft after deceased donor transplant. Also natural course of oxalate deposition in allograft kidney, coronary artery calcification, and retinal deposition after transplantation were documented.
CASE PRESENTATION
A 38-year-old male patient with history of recurrent nephrolithiasis developed end-stage kidney disease because of obstructive uropathy, and chronic hemodialysis treatment was started. He was diagnosed with coronary artery disease 10 years ago and has undergone coronary artery bypass surgery. After 2-year hemodialysis treatment, he received a deceased donor transplant. Prednisolone and mycophenolate mofetil (MMF) with antithymocyte globulin (3 mg/kg) as induction therapy were administered to the patient in the early posttransplantation period. The urine output was 2500–3500 mL/day during the first days of operation and there was no need for hemodialysis. The patient was reoperated on postoperative day 13 because of slowly dropping creatinine levels (4.2 mg/dL on day 13) and due to the collection surrounding the ureter. During this operation, hematoma was emptied and distal ureteric anastomosis was made and allograft kidney biopsy was performed due to late decrease in creatinine levels.
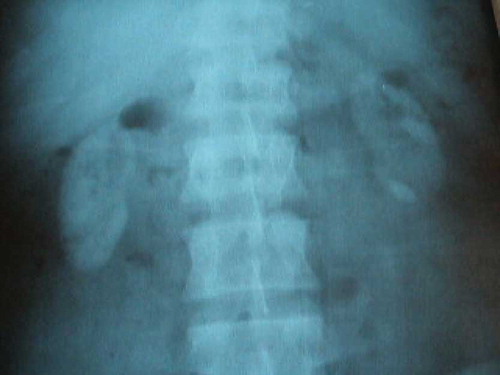
Deposition of diffuse oxalate crystal was detected in allograft kidney biopsy. Oxalate level was found to be high in a 24-hour urine specimen (118 mg/L, normal level: 7–44 mg/L). In the direct urinary system graph, the image consistent with bilateral nephrocalcinosis in the native kidneys was detected (). The patient was identified with PH and was followed up in terms of systemic oxalate deposition as well as allograft kidney.
Figure 1. The direct urinary system graphy, the image consistent with bilateral diffuse nephrocalcinosis and nephrolithiasis in the native kidneys.
Outcome of allograft kidney function, ultrasonography, and histology
The patient received maintenance immunosuppressive therapy consisting of prednisolone, mycophenolate mofetil, and tacrolimus. In the first and third months posttransplant, creatinine levels decreased to 1.4 and 1.6 mg/dL. No acute rejection episode has occurred. To prevent the formation of calcium oxalate crystals in the transplanted kidney, 10 mg/kg pyridoxine, 60 mg/kg magnesium citrate (corresponding to 10 mg/kg pure magnesium) by monitoring the blood magnesium levels, and 15–20 mg/kg phosphate were prescribed and 50 mg atenolol plus 12.5 mg hydrochlorothiazide were added to the therapy. Twenty-four-hour urine output was kept above 3–3.5 L/day. The patient's 24-hour urinary oxalate was 87 mg and blood glutamic acid level was 26.2 mg/L (normal value <13.0 mg/L).
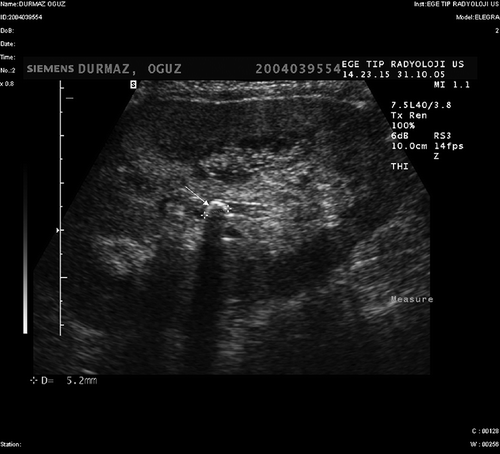
In the ultrasonography (USG) applied in the sixth month, formation of the three new calculi with the size of 5–8 mm inside the calyces, increased cortical echogenicity, and starry sky pattern composed of diffuse microcalcifications involving renal pyramids were detected (). In the control USG applied after 18 months, calculus formation disappeared and there was only a cortical echogenicity increase. Now, the patient is being followed up with a functioning graft (creatinine level 1.6 mg/dL) at 60th month.
Figure 2. The ultrasonography in the sixth month, formation of three new calculi inside the calyces, increased cortical echogenicity, and starry sky pattern composed of diffuse microcalcifications involving renal pyramids.
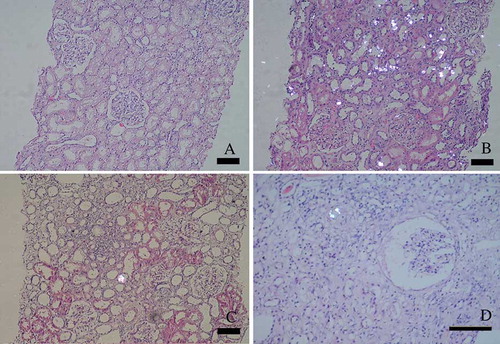
At 0-hour biopsy, glomerulus and tubular structures were normal. There were vacuolar changes in tubular epithelium. There were no oxalate crystals. Deposition of diffuse oxalate was observed on the 13th day posttransplant. At the sixth month protocol, biopsy showed a few oxalate crystals. In the kidney biopsy performed after 18 months, there were mild interstitial fibrosis and tubular atrophy with dilatation of Bowman spaces. Only a few crystals were seen ().
Figure 3. (A) Zero-hour – implantation biopsy showed only tubular vacualisation, glomerulus and interstitial structures were normal. (B) The 13th posttransplantation day allograft biospy showed intensive oxalate crystals deposition. (C) The sixth month renal biopsy showed a few oxalate crystals. (D) The last biopsy was done on day 497. Renal allograft biopsy showed mild interstitial fibrosis/tubular atrophy with dilatation of Bowman spaces. Only a few crystals were seen (bar length 100 nm).
Ocular findings
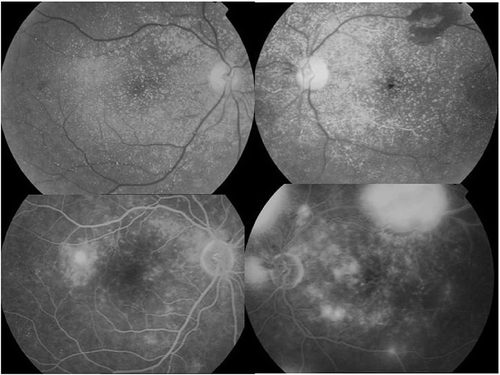
In the fundus examination preretinal, intraretinal, and intravascular diffuse oxalate crystals were detected in both eyes, especially in the posterior pole (). In the left eye, diffuse soft exudates and a large preretinal hemorrhage on the arch of the upper temporal vessel were observed. Fundus fluorescein angiography (FFA) showed an extrafoveal choroidal neovascular membrane (CNV) temporal to the macula in the right eye and a large ischemia and retinal neovascularization in the left eye. CNV in the right eye was treated by argon laser photocoagulation. Left eye was given panretinal photocoagulation to regress retinal neovascularization. FFA was repeated after 3 weeks to evaluate the efficacy of the treatment. Because neovascularization leakage was active, increased laser was applied to the left eye, and in the right eye, laser was repeated because active leakage of CNV had been lasting. In the ninth month examination, crystals stayed the same, but there was a significant decrease in leakage in the CNV region in the right, and neovascularization in the left was inactive. In the 18th month retina examination and angiography, oxalate crystals were found to be significantly regressed ().
Figure 4. In the fundus examination preretinal, intraretinal, and intravascular diffuse oxalate crystals were detected on both eyes, especially dense in the posterior pole; in the left eye, diffuse soft exudates and a large preretinal hemorrhage on the the upper temporal vessel arch.
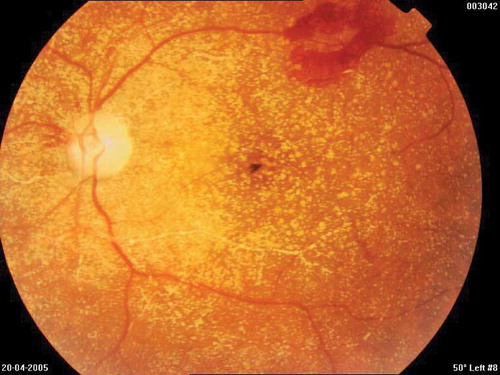
Figure 5. In the fundus examination, in the bottom two pictures, preretinal, intraretinal, and intravascular diffuse oxalate crystals decrease after 18th month.
Cardiovascular findings
In the multislice CT images, diffuse calcifications were observed in left anterior descending, left circumflex, and right coronary arteries, and total Agatston score was calculated as 1163. These findings revealed that the plaque load was high and the risk of coronary artery stenosis causing more than 50% shrinkage in diameter was high. Contrast coronary angiography examination was not made due to excessive calcifications in the patient. In the multislice CT repeated after the 18th month, same findings were found.
DISCUSSION
PH1 is an autosomal recessive disease resulting from a defect in glyoxylate metabolism which depends on the lack or absence of AGT activity, a liver-specific peroxisomal enzyme. Thus, urinary excretion of oxalate and glyoxylate increases. The disease concomitant with massive intrarenal Ca oxalate deposits was identified as PH in 1957. PH2, however, results from glyoxylate reductase (GR) and hydroxy pyruvate reductase enzyme defects.Citation3–6 Both disorders are characterized by hyperoxaluria. In addition, there is increased urinary excretion of glycolate in PH1, while there is increased urinary excretion of l-glyceric acid in PH2.Citation2
Death without treatment occurs in the second and third decades. There are three transplantation options for patients with PH1: (1) isolated kidney transplantation; (2) isolated liver transplantation to repair the metabolic defect before significant kidney damage occurs; and (3) combined hepatorenal transplantation.Citation7,Citation8
Early kidney transplantation is evidently sufficient to eliminate oxalate deposits at least partially and has the effectiveness to improve patient's condition without liver transplantation.Citation9 This option should be considered in pyridoxine-sensitive patients even in the existence of systemic oxalosis because of the good results of isolated kidney graft.Citation10,Citation11
Allen et al. identified a patient with clinical and biochemical existence of residual AGT activity and with a perfect renal function without the formation of a new calculus 1 year after renal transplantation from a live donor. In this woman, pyridoxine-sensitive PH1 was diagnosed after the isolated kidney transplantation. The renal function was improved after adding pyridoxine and it was still at a satisfactory level after 42 months.Citation12 Kamal Sud et al. showed the recovery of pancytopenia after successful single kidney transplantation in their case. They reported that a good functioning kidney graft affects systemic oxalate load in a positive way and heals systemic complications of oxalosis at least for a period of time. Although the outcome of deceased donor kidney transplantation is inferior, the results of live donor kidney transplantation are promising.Citation2
In our case, nephrocalcinosis, nephrolithiasis, and coronary artery disease due to diffuse calcification in coronary arteries of the patient strongly suggested the presence of oxalosis. But, definite diagnosis was made at the 13th day allograft biopsy. Repeated biopsy samples showed the crystal deposition decreased in intensity. During posttransplant period preventive measures including pyridoxine, magnesium, hydrochlorothiazide, and fluid intake were intensively applied. The aim of this approach is not only to cease kidney involvement but also to prevent the progressive nature of oxalate deposition in other organ and soft tissues.Citation8,Citation9,Citation13,Citation14 In this respect retinal examination gives opportunity to evaluate oxalate deposition. Small et al. presented 24 patients with ocular involvement. Eight out of 24 patients have bilateral symmetric retinopathy. Existence of maculopathy in cases was accompanied by more severe systemic involvement for oxalosis.Citation15 In our case, bilateral preretinal, intraretinal, and intravascular diffuse oxalate crystals were detected, and argon laser photocoagulation treatments were needed for choroidal and retinal neovascularization. Repeated examinations showed the regressive nature of oxalate depositions.
Our patient probably had PH1 because of the existence of nephrocalcinosis and systemic involvement, both of which are extraordinary findings in PH2. This comment is only a thought because there is no enzymatic study on liver biopsy specimen.
The ideal therapy for patients with end-stage renal failure secondary to PH and with systemic oxalosis is liver and kidney transplantation together, because a functioning liver provides replacement of missing enzyme and a functioning kidney provides excretion of oxalate crystals. However, it is suggested that isolated kidney transplantation may be a therapeutic option to slow down the systemic effects of oxalosis.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper.
REFERENCES
- Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU. Renal allograft survival in patients with oxalosis. Transplantation. 2002;74(5):707–710.
- Sud K, Swaminathan S, Varma N, Reversal of pancytopenia following kidney transplantation in a patient of primary hyperoxaluria with bone marrow involvement. Nephrology. 2004;9:422–427.
- Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12:1986–1993.
- Scheid C, Koul H, Hill WA, Oxalate toxicity in LLC-PK1 cells: Role of free radicals. Kidney Int. 1996;49:413–419.
- Levin-Iaina N, Dinour D, Romero L, Late diagnosis of primary hyperoxaluria type 2 in the adult: Effect of a novel mutation in GRHPR gene on enzymatic activity and molecular modeling. J Urol. 2009;181(5):2146–2151.
- Scheinman JI. Recent data on results of isolated kidney or combined kidney/liver transplantation in the U.S.A. for primary hyperoxaluria. J Nephrol. 1998;11(Suppl. 1):42–45.
- Ellis SR, Hulton SA, McKiernan PJ, de Ville de Goyet J. Combined liver–kidney transplantation for primary hyperoxaluria type 1 in young children. Nephrol Dial Transplant. 2001; 16:348–354.
- Cochat P. Primary hyperoxaluria type 1. Kidney Int. 1999;55: 2533–2547.
- Cochat P, Gaulier JM, Koch Nogueria PC, Combined liver-kidney transplantation in primary hyperoxaluria type 1. Eur J Pediatr. 1999;158(Suppl. 2):S75–S80.
- Marangella M. Transplantation strategies in type 1 primary hyperoxaluria: The issue of pyridoxine responsiveness. Nephrol Dial Tansplant. 1999;14:301–303.
- Marangella M, Vitale C, Petrarulo M, Bony content of oxalate in patients with primary hyperoxaluria or oxalosis unrelated renal failure. Kidney Int. 1995;48:182–187.
- Allen AR, Thompson EM, Williams G, Watts RWE, Pusey CD. Selective renal transplantation in primary hyperoxaluria type 1. Am J Kidney Dis. 1996;27:891–895.
- Chetyrkin SV, Kim D, Belmont JM, Scheinman JI. Pyridoxamine lowers kidney crystals in experimental hyperoxaluria: A potential therapy for primary hyperoxaluria. Kidney Int. 2005;67:53–60.
- Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long-term treatment with orthophosphate and pyridoxine in patient with primary hyperoxaluria. N Engl J Med. 1994;331:1553–1558.
- Small KW, Letson R, Scheinman J. Ocular finding in primary hyperoxaluria. Arch Ophthalmol. 1990;108(1):89–93.