Abstract
Common variable immunodeficiency disease (CVID) represents a heterogeneous group of primary hypogammaglobulinemias of unknown etiology, characterized by decreased serum immunoglobulin levels and recurrent bacterial infections and is often accompanied by autoimmune disease. Renal involvement is rare in CVID, despite widespread involvement of other organ systems. We describe a 21-year-old girl who presented with recurrent infections, hepatosplenomegaly, renomegaly, and renal insufficiency. Renal biopsy revealed a remarkable diffused interstitial infiltration and severe degenerative tubular lesions. Interstitial infiltration consisted mainly of CD8+ T cells and CD68+ macrophages with less CD4+ T and rare B cells. For the cases with recurrent infections, multiple organomegaly, and renal insufficiency, clinicians should consider to exclude CVID, so as to make the timely diagnosis and appropriate management.
INTRODUCTION
Common variable immunodeficiency disease (CVID) represents a heterogeneous group of primary hypogammaglobulinemias of unknown etiology, characterized by decreased serum immunoglobulin levels and recurrent bacterial infections and is often accompanied by autoimmune disease. Accumulating data suggest that T-cell and monocyte abnormalities may lead to a defect in B-cell maturation and function and a lack in antigen-specific T cells. Renal involvement is rare in CVID, despite widespread involvement in other organ systems. We describe a patient who developed notable renomegaly with severe renal interstitial infiltration and renal insufficiency in CVID.
CASE REPORT
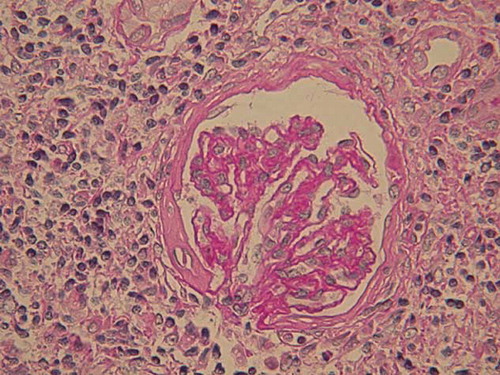
A 21-year-old girl presented with a story of recurrent infections, including upper respiratory tract infection, septicemia, and pneumonia, as well as herpes and otitis media. She was known to have persistent hepatosplenomegaly since 11 years, when she was hospitalized with septicemia. As the serum liver tests were normal then, no further examination was performed; however, she got well eventually after given antibiotics and intravenous immunoglobulin. From then on, she accepted antibiotics intermittently, but no immunoglobulin infusion or any other therapy. In recent 3 months, she gradually developed increasing fatigue, abdominal distension, anorexia, and frequent urination. No special family history was available. Routine physical examination indicated the following signs: blood pressure 115/70 mmHg, appearance of anemia, hepatomegaly, splenomegaly. Laboratory tests showed serum creatinine 164.0 μmol/L (normal 53–115 μmol/L); blood urea nitrogen 13.2 mmol/L (normal 1.43–7.14 mmol/L); hemoglobin 84 g/L; mean globular volume 74 fL; leukocytes 3.68 × 109/L; lymphocytes 1.72 × 109/L; and platelets 144 × 109/L. Liver tests showed alanine transaminase 50 U/L, glutamic-oxaloacetic transaminase 68 U/L, alkaline phosphatase 910 U/L, γ‐glutamyltransferase 421 U/L, albumin 37.8 g/L, globulin 24.8 g/L (normal 20–35 g/L). Urinalysis showed hyposthenuria, specific gravity 1.010, with negative proteinuria (<0.15 g/day), no hematuria or leukocyturia. Serologic tests for human immunodeficiency virus, hepatitis C virus, hepatitis B virus, as well as hepatitis C virus RNA and hepatitis B virus DNA assays were negative. Serum cytomegalovirus DNA and Epstein–Barr virus DNA were undetectable. Levels of complement C3 and C4 components were normal. Antinuclear antibodies, anti-Sm antibodies, and anti-extractable nuclear antibodies, as well as antithyroglobulin, antimitochondrial, and anti-neutrophil cytoplasmic autoantibodies were undetectable. Immunoglobulin levels were dramatically decreased, including IgG 2.13 g/L (normal 7.23–16.85 g/L); IgA < 0.25 g/L (normal 0.69–3.85 g/L); IgM < 0.17 g/L (normal 0.63–2.77 g/L). Immunological investigations showed normal lymphocyte subpopulations, with normal T lymphocyte (CD3+, CD4+, and CD8+ cells, CD4+/CD8+ ration), B lymphocyte (CD19+ cells), and NK lymphocyte (CD16+ + 56+) counts. Her karyotype was normal, with an absence of abnormal DNA breakage in lymphocytes. Anemia-related serum tests showed serum iron 3.30 μmol/L (normal 10.7–26.9 μmol/L); total iron binding capacity 72.2 μmol/L (normal 48.3–68 μmol/L); ferritin 23.8 ng/mL (normal 100–300 ng/mL); folic acid 6.91 ng/mL; vitamin B12 275 pg/mL. Plasma thyroid hormone, cortisol, and gonadal hormone levels were normal. A chest X-ray remained normal. The MRI brain scan found a microadenoma of pituitary with abscess and necrosis. Abdominal ultrasonography revealed hepatomegaly (maximal anteroposterior diameter 13.64 cm for right lobe, maximal oblique diameter 16.6 cm), splenomegaly, as well as notable renomegaly (16.5 × 5.1 cm, 15.1 × 5.8 cm, separately) with increased echo (). Gastroscope examination indicated chronic superficial gastritis with Helicobacter pylori test negative. Additional biopsies were performed. Bone marrow biopsy showed obvious proliferation and iron deficiency anemia. Liver biopsy revealed portal area enlargement and mild polyclonal T (CD3+) lymphocyte infiltration. Histologic examination of a renal biopsy sample revealed a remarkable diffused interstitial infiltration and severe degenerative tubular lesions (). Interstitial infiltration consisted mainly of CD8+ T cells and CD68+ macrophages with less CD4+ T cells and rare B cells. Glomeruli were found with different degrees of ischemic change or sclerosis (). Vessels were normal. Immunohistochemical staining for hepatitis B surface antigen and hepatitis B core antigen and immunofluorescence staining for IgA, IgG, IgM, C3, C1q, and Fib were negative. Based on all the above examinations, the patient was diagnosed with CVID, renal insufficiency, iron deficiency anemia, and pituitary microadenoma. Additionally, she underwent upper respiratory infection and periodontitis with intermittent fever during hospitalization. As soon as the diagnoses were established, this patient was referred to the local hospital voluntarily for monthly intravenous immunoglobulin management on account of the medical insurance. The follow-up in telephone call shows she has no recurrence of infections, with a stable creatinine of 200 μmol/L 6 months after her discharge.
Figure 1. Abdominal ultrasonography revealed notable renomegaly with increased echo, the right kidney taken for example.
Figure 2. Light microscopy of renal tissues with remarkable diffused interstitial infiltration and severe degenerative tubular lesions (periodic acid-Schiff stain, original magnification ×100).
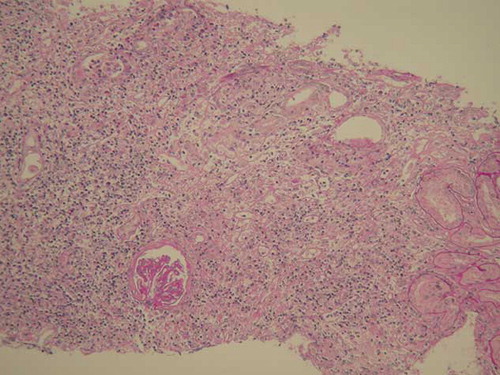
Figure 3. Light microscopy of a glomerulus with ischemic change as well as areas of interstitial infiltration and severe degenerative tubular lesions (periodic acid-Schiff stain, original magnification ×400).
DISCUSSION
CVID was first described in 1953 and is the most common primary immunodeficiency disease. The incidence of CVID ranges from 1/25,000 to 1/66,000 inhabitants, though the milder cases probably go undetected. The age at the onset of symptoms was 2–66 years, 33% patients younger than 14 years of age, with no difference of incidence between males and females.Citation1 Our patient had a history of recurrent infections and dramatically decreased immunoglobulin levels – two highly suggestive features of CVID. Female sex and the presence of circulating B lymphocytes exclude the diagnosis of the common form of X-linked agammaglobulinemia. Thus, the diagnosis of CVID in our patient was made according to the ESID/PAGID criteria.Citation2 CVID is an idiopathic group of disorders with multiple systems involved, and the clinical manifestations are diverse. Recurrent infections are the most prominent clinical problems observed in patients with CVID, including respiratory tract infections, pneumonia, otitis, sinusitis, as well as diarrhea and sepsis. Hepatosplenomegaly and lymphoid hyperplasia are also common as well as autoimmune disease. Granulomatous disease occurs in 9% of patients with CVID and commonly involves the liver, spleen, lymph nodes, lung, skin, and bone marrow.Citation3 Cancers develop in 6% of patients, and all lymphomas are B-cell type. Because of multiplex clinical manifestation, failing to diagnose and misdiagnose of CVID are frequent. It was reported that the mean diagnostic time was delayed for 8.9 years.Citation1 Our patient suffered from recurrent infections and hepatosplenomegaly, but did not have cancers and autoimmune disease yet. Iron deficiency anemia may be associated with her poor appetite and chronic superficial gastritis. And the diagnosis of CVID was made 11 years after the clinical onset.
Renal involvement seems rare. It was noted in only 5 of 240 CVID patients with no further available details.Citation4 Miller et al.Citation5 reported CVID in a renal transplant patient presumably resulting from long-standing immunosuppression. Hogan et al.Citation6 described a renal transplant patient with preexisting CVID. The renal pathology before transplantation showed chronic interstitial changes and nephrosclerosis consistent with end-stage renal disease. However, the relationship between CVID and renal lesions was not discussed. Both of the two patients experienced severe infections and allograft rejections. Interestingly, despite the presence of a primary T-cell immunodeficiency in these patients, allograft biopsy showed cellular rejection.Citation6 There have been three previous case reports of renal interstitial granulomas in CVID.Citation3,Citation7,Citation8 In one of the cases, there was associated immune complex deposition, and the patient presented with acute renal failure 17 years after the initial diagnosis of CVID.Citation8 Besides, the interstitial nephritis with predominantly CD8+ T lymphocyte infiltration was also reported.Citation9 In all the cases with kidney involved, although interstitial nephritis, granulomas, or immune complex glomerulonephritis were described, renomegaly was not found. Our patient presented with renomegaly and renal insufficiency with unknown reason. Renal biopsy disclosed remarkable renal interstitial infiltration and severe tubular lesions, which were in accordance with the clinical features.
The pathogenesis of renal injury in CVID is unclear. It seems that nonspecific polymorphous proliferation in bone marrow was not related to such interstitial infiltration in this patient. Poor proliferations of T-cell to mitogens and decreased CD4+/CD8+ ratio have been reported in CVID patients presenting with granulomatous disease.Citation10 In our patient, although the CD4+/CD8+ ratio in peripheral blood was normal, CD8+ T cells occupied a position of prominence in the interstitial infiltration cells. It has been shown in vitro that there is upregulated monocyte production of interleukin 12 (IL-12), leading to increased production of interferon-γ by T lymphocytes.Citation11 Tumor necrosis factor-α (TNF-α), a proinflammatory cytokine involved in granuloma formation, has been proved to be persistently activated in patients with granulomatous CVID.Citation12 Thus, we carried out additional immunohistochemical staining with spared renal specimen and found enhanced expression for TNF-α and IL-12 in interstitial cells, but negative for interferon-γ. Altogether, T-cell abnormalities and upregulated monocyte production proinflammatory may play a vital role in CVID patients, and finally induce renal injury, such as tubular degeneration and glomeruli ischemia. Anyway, as the documented cases are limited, further research should be done to elucidate the underlying pathological mechanisms of renal involvement in CVID.
There is no complete cure. Long-term administration of immunoglobulins reduces the incidence of infections, even in renal transplant patients with appropriate immunosuppression therapy.Citation5,Citation6 GlucocorticosteroidCitation7 or glucocorticosteroid combined with rituximabCitation13 may be effective in cases with massive proteinuria and progressive renal failure. In conclusion, CVID is a congenital group of systemic disorders with varied manifestations. For the cases represented by recurrent infections, multiple organomegaly, and renal insufficiency, clinicians should consider to exclude CVID, so as to make the timely diagnosis and appropriate management.
Declaration of interest:
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Quinti I, Soresina A, Spadaro G, Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27(3):308–316.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan‐American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190–197.
- Fakhouri F, Robino C, Lemaire M, Granulomatous renal disease in a patient with common variable immunodeficiency. Am J Kidney Dis. 2001;38(2):E7.
- Hermaszewski RA, Webster AD. Primary hypoglobulinaemia: A survey of clinical manifestations and complications. Q J Med. 1993;86:31–42.
- Miller BW, Brennan DC, Korenblat PE, Goss JA, Flye MW. Common variable immunodeficiency in a renal transplant patient with severe recurrent bacterial infection: A case report and review of the literature. Am J Kidney Dis. 1995;25(6):947–951.
- Hogan MB, Wilson NW, Muchant DG. Renal transplantation in a patient with common variable immunodeficiency. Am J Kidney Dis. 1999;33(6):e7.
- Meyer A, Lachmann HJ, Webster AD, Burns A, Thway K. Hypercalcemia in a patient with common variable immunodeficiency and renal granulomas. Am J Kidney Dis. 2005;45(5): e90–e93.
- Stigant C, Sapir D, Sweet J, Downey G, Bargman JM. A unique renal lesion in common variable immunodeficiency. Clin Nephrol. 2002;57(1):74–79.
- Kano H, Sugamoto K, Goto M, A case of common variable immunodeficiency with intractable diarrhea and the functional disorder of renal tubules. Nihon Rinsho Meneki Gakkai Kaishi. 2000;23(2):163–172.
- Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–617.
- Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: A fundamental defect in common variable immunodeficiency. J Immunol. 2000; 164:488–494.
- Aukrust P, Lien E, Kristoffersen AK, Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency-possible immunologic and clinical consequences. Blood. 1996;87:674–681.
- Benoit G, Lapeyraque AL, Sartelet H, Saint-Cyr C, Le Deist F, Haddad E. Renal granuloma and immunoglobulin M‐complex glomerulonephritis: A case of common variable immunodeficiency? Pediatr Nephrol. 2009;24(3):601–604.