Abstract
We reported a case of a 41-year-old woman who had been diagnosed with Gitelman’s syndrome since the age of 31 years. The diagnosis was established by the typical biochemical pictures including renal wasting hypokalemia, hypomagnesemia, hypocalciuria, metabolic alkalosis, and hyperreninemic hyperaldosteronism. She had normal blood pressure and had never used diuretics. She had a sibling with similar syndrome. The patient was treated with oral potassium and magnesium supplementation. She began to have hypercalcemia at the age of 39 years. The diagnostic approach to hypercalcemia became more complicated because of normal parathyroid hormone levels and underlying hypocalciuria due to Gitelman’s syndrome. Thorough evaluation eventually identified primary hyperparathyroidism as the cause of hypercalcemia. To our best knowledge, this is the first report of combined occurrence of Gitelman’s syndrome and primary hyperparathyroidism in the literature.
INTRODUCTION
Gitelman’s syndrome is a rare autosomal recessive salt-losing tubulopathy of young adults caused by a mutation of genes encoding the human sodium chloride cotransporter and magnesium channels in the thiazide-sensitive segment of the distal convoluted tubule.Citation1 People suffering from Gitelman’s syndrome display many of the biochemical changes which are identical to those of patients who are on thiazide diuretics.Citation2 Indeed, the surreptitious abuse of diuretics remains the commonest differential diagnosis. The biochemical pictures are characterized by renal wasting hypokalemia, hypomagnesemia, hypocalciuria, metabolic alkalosis, and hyperreninemic hyperaldosteronism.Citation3 Many symptoms are related to these electrolyte abnormalities. However, unlike thiazide diuretic use, Gitelman’s syndrome has never been reported to be associated with hypercalcemia. Herein, we reported an unusual case of Gitelman’s syndrome complicated with hypercalcemia.
CASE REPORT
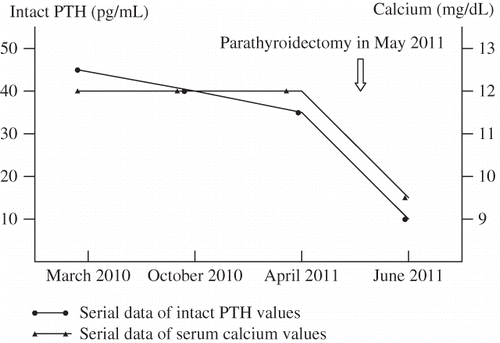
A 41-year-old woman was referred to nephrologic clinic for evaluation of persistent hypercalcemia. She had been diagnosed with Gitelman’s syndrome since the age of 31 years. The diagnosis of Gitelman’s syndrome was established by the typical biochemical pictures including renal wasting hypokalemia, hypomagnesemia, hypocalciuria, metabolic alkalosis, and hyperreninemic hyperaldosteronism. She had normal blood pressure and had never used diuretics. She had a sibling with similar syndrome. The patient was treated with oral potassium and magnesium supplementation and received regular follow-up at outpatient clinic. However, the restoration of normal potassium and magnesium balance had been difficult to achieve, with serum potassium levels ranging from 2.0 to 2.6 mmol/L (normal range 3.5–5.0 mmol/L) and serum magnesium levels ranging from 1.1 to 1.7 mg/dL (normal range 1.9–2.5 mg/dL). Hypercalcemia was first detected incidentally when she was aged 39 years, with a serum calcium level of 10.8 mg/dL (normal range 8.9–10.3 mg/dL). Further investigations included ionized calcium 1.32 mmol/L (normal range 0.9–1.25 mmol/L), phosphate 2.2 mg/dL (normal range 2.5–4.5 mg/dL), intact parathyroid hormone (PTH) 45.9 pg/mL (normal range 12–65 pg/mL, using immunometric two-site sandwich assays), and 25-dihydroxyvitamin D 16.9 ng/mL (sufficient >25 ng/mL, toxic >100 ng/mL). Urinary calcium excretion was 45 mg/24 h. Her thyroid function was normal. Protein electrophoresis did not disclose monoclonal gammopathy in her serum or free light chain in her urine. Assays for PTH-related peptide and 1,25-dihydroxyvitamin D were unavailable at our institution. She denied treatment with thiazide, lithium, calcium supplements, vitamin D, or vitamin A. The patient had persistent hypercalcemia and hypophosphatemia in the course of 2-year follow-up, with serum calcium levels ranging from 11.0 to 12.1 mg/dL (normal range 8.9–10.3 mg/dL) and serum phosphate levels ranging from 2.1 to 2.4 mg/dL (normal range 2.4–4.7 mg/dL). Intact PTH was rechecked once in the meantime and was still within the normal range (38.9 pg/mL, normal range 16–87 pg/mL). The patient was referred because of unexplained hypercalcemia.
We reviewed previous laboratory data and thought that the PTH levels were still inappropriately high considering the presence of hypercalcemia. Therefore, primary hyperparathyroidism was still suspected. A neck ultrasonography revealed a hypoechoic lesion 1.0 × 1.0 cm at the upper pole of right lobe of thyroid and a hypoechoic lesion 1.5 × 0.8 cm at the lower pole of left lobe of thyroid. A radionuclide parathyroid scan (technetium-99m MIBI) showed areas of increased uptake in the same regions. A classic neck exploration with a horizontal thyroid incision was performed. Histological study confirmed the diagnosis of parathyroid adenoma. Serum calcium returned to normal levels immediately after surgical removal of the parathyroid adenomas ().
Figure 1. Serial data of intact PTH and serum calcium value.
DISCUSSION
Thiazide diuretics inhibit sodium chloride cotransporter in the distal convoluted tubule. Hypercalcemia associated with thiazide diuretic use is a well-recognized clinical entity.Citation4 Thiazides have several metabolic effects that may contribute to increased calcium levels. A reduction in urine calcium excretion is the most likely explanation.Citation5,6 It has been shown that thiazide-induced hypocalciuria occurred only when the diuretic induced hypovolemia.Citation7 The extracellular volume contraction would activate the sympathetic nervous system and promote a proximal sodium and water hyper-absorption that would induce an electrostatic intraluminal positive potential driving a paracellular proximal calcium absorption. Metabolic alkalosis associated with diuretic use could also cause an elevation in total serum calcium through a pH-dependent increase in protein-bound calcium. Although plasma 1,25-dihydroxyvitamin D levels are unchanged, increased intestinal calcium absorption in response to thiazide treatment has been noted and could also contribute to an increase in serum calcium.Citation8 A final factor that may lead to the development of hypercalcemia is hemoconcentration associated with diuresis.Citation9
Gitelman’s syndrome is caused by a mutation of genes encoding the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Patients with Gitelman’s syndrome present some clinical and biochemical alterations resembling that observed in thiazide diuretic use. However, Gitelman’s syndrome has never been reported to be associated with hypercalcemia. Hypomagnesemia may be the major cause. Hypomagnesemia can impair the function of calciotropic hormones. The inverse relationship between plasma-ionized calcium, PTH, and calcitriol was found to be downregulated in patients with Gitelman’s syndrome suggesting that they have a reduced skeletal sensitivity to PTH and an impaired intestinal calcium transport in spite of normal plasma calcitriol levels.Citation10 This blunted response may also explain the lack of hypercalcemic response to hypocalciuria in patients with Gitelman’s syndrome and the recovery of the normal correlation between plasma-ionized calcium and calciotropic hormones after plasma magnesium correction.Citation11 The hypomagnesemia-induced lower intestinal and skeletal sensitivity to the calciotropic hormones is further confirmed by the different behaviors of calcium pool observed in chronic thiazide-treated subjects and in Gitelman’s syndrome patients. In the former group, hypocalciuria is followed by a calcium pool expansion with an increase of the calcium bone content; whereas in patients with Gitelman’s syndrome, hypocalciuria does not induce any change of the bone mineral content.Citation10
Therefore, the presence of hypercalcemia in the course of Gitelman’s syndrome is extremely unusual and requires thorough evaluation. However, the diagnostic approach to hypercalcemia became more complicated because of normal PTH levels and underlying hypocalciuria due to Gitelman’s syndrome. In the present case, the “normal” PTH values in the presence of hypercalcemia are inappropriately high; and patients with non-parathyroid hypercalcemia virtually always have serum PTH values below 20–25 pg/mL (normal range 10–60 pg/mL).Citation12 Although the diagnosis of primary hyperparathyroidism is straightforward in most patients, approximately 10–20% of patients exhibit only minimally elevated or normal serum PTH values, ranging from 35 to 65 pg/mL.Citation13,14 Therefore, primary hyperparathyroidism should still be suspected in our patient. However, in this circumstance, the diagnosis of familial hypocalciuric hypercalcemia also should be considered. Approximately 15–20% of patients with familial hypocalciuric hypercalcemia may have mildly elevated serum PTH values due to calcium-sensing receptor mutation causing the parathyroid glands to be less sensitive to serum calcium, resulting in normal to high PTH despite elevated serum calcium levels.Citation15
It is important to make a differential diagnosis between primary hyperparathyroidism and familial hypocalciuric hypercalcemia because the latter is a benign condition that does not require parathyroidectomy. The major feature that distinguishes familial hypocalciuric hypercalcemia from primary hyperparathyroidism is a low urinary calcium excretion in the former. Unfortunately, it is unhelpful in our case because of underlying hypocalciuria related to Gitelman’s syndrome. Nevertheless, primary hyperparathyroidism was still preferred due to other clinical information including hypophosphatemia and findings of neck ultrasonography and radionuclide parathyroid scan.
The cause of low or normal intact PTH levels in the case of primary hyperparathyroidism is uncertain. There are several potential reasons. First, intact PTH is a heat-labile and fragile peptide that degrades rapidly and may result in spuriously low intact PTH levels if the cold chain is not maintained during sample collection and transportation.Citation16 However, the intact PTH was measured on more than two occasions with similar results in our patient. Second, coexistent sarcoidosis or vitamin D toxicity might suppress intact PTH levels even in patients with primary hyperparathyroidism and hypercalcemia,Citation17 but neither clinical problem was present in our patient. Third, another uncommon cause for low intact PTH levels is hypomagnesemia because magnesium is necessary for both synthesis and secretion of intact PTH.Citation18 Therefore, hypomagnesemia may be the major cause in our patient.
In conclusion, we reported a case of a Gitelman’s syndrome patient with sustained hypercalcemia, normal PTH levels, and surgically proven primary hyperparathyroidism. The combined occurrence of Gitelman’s syndrome and primary hyperparathyroidism may be coincident. This case indicated that a normal serum PTH value in the setting of hypercalcemia still suggests the diagnosis of primary hyperparathyroidism. In patients with Gitelman’s syndrome, it is difficult to distinguish primary hyperparathyroidism from familial hypocalciuric hypercalcemia because of underlying hypocalciuria.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
REFERENCES
- Scheinman SJ, Guay-Woodford LM, Thakker RV, Warnock DG. Genetic disorders of renal electrolyte transport. N Engl J Med. 1999;340:1177–1187.
- O’Shaughnessy KM, Karet FE. Salt handling and hypertension. J Clin Invest. 2004;113:1075–1081.
- Graziani G, Fedeli C, Moroni L, Cosmai L, Badalamenti S, Ponticelli C. Gitelman syndrome: Pathophysiological and clinical aspects. QJM. 2010;103:741–748.
- Wermers RA, Kearns AE, Jenkins GD, Melton III LJ. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am J Med. 2007;120(911):e9–e15.
- Brickman AS, Massry SG, Colburn JW. Changes in serum and urinary calcium during treatment with hydrochlorothiazide: Study on mechanisms. J Clin Invest. 1972;51:945–954.
- Middler S, Pak CY, Murad F, Bartter FC. Thiazide diuretics and calcium metabolism. Metabolism. 1973;22:139–146.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J,Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658.
- Bazzini C, Vezzoli V, Sironi C, . Thiazide-sensitive NaCl-cotransporter in the intestine: Possible role of hydrochlorothiazide in the intestinal Ca2+ uptake. J Biol Chem. 2005;280:19902–19910.
- Heath III H. Postural and venous stasis-induced changes in total calcium. Mayo Clin Proc. 2005;80:1101.
- Bianchetti MG, Bettinelli A, Casez JP, . Evidence for disturbed regulation of calciotropic hormone metabolism in Gitelman syndrome. J Clin Endocrinol Metab. 1995;80:224–228.
- Bettinelli A, Basilico E, Metta MG, Borella P, Jaeger P, Bianchetti MG. Magnesium supplementation in Gitelman syndrome. Pediatr Nephrol. 1999;13:311–314.
- Grant FD, Conlin PR, Brown EM. Rate and concentration dependence of parathyroid hormone dynamics during stepwise changes in serum ionized calcium in normal humans. J Clin Endocrinol Metab. 1990;71:370–378.
- Nussbaum SR, Zahradnik RJ, Lavigne JR, . Highly sensitive two-site immunoradiometric assay of parathyrin, and its clinical utility in evaluating patients with hypercalcemia. Clin Chem. 1987;33:1364–1367.
- Silverberg SJ. Diagnosis, natural history, and treatment of primary hyperparathyroidism. Cancer Treat Res. 1997;89:163–181.
- Fuleihan Gel H. Familial benign hypocalciuric hypercalcemia. J Bone Miner Res. 2002;17(2):N51–N56.
- Holmes DT, Levin A, Forer B, Rosenberg F. Preanalytical influences on DPC IMMULITE 2000 intact PTH assays of plasma and serum from dialysis patients. Clin Chem. 2005;51:915–917.
- Kinoshita Y, Taguchi M, Takeshita A, Miura D, Tomikawa S, Takeuchi Y. 1,25-Dihydroxyvitamin D suppresses circulating levels of parathyroid hormone in a patient with primary hyperparathyroidism and coexistent sarcoidosis. J Clin Endocrinol Metab. 2005;90:6727–6731.
- Rude RK. Magnesium deficiency in parathyroid function. In: Bilzekian JP, Marcus R, Levine MA, eds. The Parathyroids. New York: Raven Press; 2001:763–777.