Abstract
A 47-year-old man was admitted to hospital for migratory joint pain, fatigue, and cough with bloody sputum and proteinuria with increased serum creatinine level. Diagnosis of Wegener’s granulomatosis was established. During follow-up, the vena cava superior syndrome developed. The patient died of respiratory failure after 12 years of follow-up. The autopsy revealed rigid, whitish, 12 mm thick tissue, which embedded and compressed the large vessels upwards from their origin in the heart, thus causing vena cava superior syndrome. This tissue was composed of fibrous material without inflammatory cellulization. We consider this fibrous tissue as a manifestation of fibrosing mediastinitis that may or may not share pathogenesis with Wegener’s granulomatosis.
INTRODUCTION
Wegener’s granulomatosis belongs to a group of ANCA-positive vasculitides. Typically, it manifests as an upper respiratory tract involvement resistant to antibiotic treatment, which is frequently followed by a renal lesion of a rapidly progressive glomerulonephritis type. Any other organ system may be afflicted, too. We present a case of a 47-year-old man with Wegener’s granulomatosis that displayed vena cava superior syndrome due to the compression of the vena cava by masses of fibrous tissue.
CASE
A 47-year-old man with an unremarkable medical history was referred to our hospital in March 1989 because of migratory joint pain, fatigue, and cough with bloody sputum. The disease did not respond to antibiotic treatment. After 3 weeks, proteinuria 6.2 g/24 h, hematuria 800,000 erythrocytes/min, serum creatinine level 247 μmol/L, and creatinine clearance 0.42 mL/s were documented. Serum c-ANCA was positive. Circulating immune complexes, serum immunoglobulins, C3 and C4 complement components were normal. A chest X-ray displayed infiltrates in both lungs with no apparent granulomas. The spirometry revealed restrictive respiratory disorder. A damaged diffusion capacity of the lungs was also demonstrated. The kidney and lung biopsies established the diagnosis of Wegener’s granulomatosis, with focal and segmental pauci-immune intra-extracapillary necrotizing glomerulonephritis, stenosing arteriolar infiltrates in the kidney tissue and giant cell granulomatous vasculitis in the lungs. After a 6-week treatment with cyclophosphamide 150 mg/day and prednisone 30 mg/day, the patient’s complaints vanished, proteinuria decreased to 1 g/24 h, serum creatinine level was 127 μmol/L, and creatinine clearance 1.27 mL/s. ANCA turned negative. Cyclophosphamide treatment was stopped and the patient was taking only prednisone in a dose gradually decreased to 10 mg every other day till December 1990 (21 months), when Wegener’s granulomatosis relapsed.
The patient suffered again from cough with bloody sputum, fatigue, E.S.R. was 150/h, anemia with hemoglobin 70 g/L, proteinuria 5.7 g/24 h, creatinine clearance was 1.28 mL/s, and ANCA was again positive. A chest X-ray revealed a single lung granuloma. Pulse methylprednisolone in a dose of 10 × 250 mg followed by prednisone (25 mg/day) and cyclophosphamide 150 mg/day for 1 month and then chlorambucil 5 mg/day were administered. After 1 month, the cough subsided, proteinuria decreased to 1.6 g/24 h, creatinine clearance was normal, and ANCA disappeared. We continued the administration of chlorambucil 2.5 mg every other day for 1 year and prednisone in a dose decreasing to 2.5 mg every other day for 5 years.
The second relapse appeared in July 1997 (5 years after chlorambucil and 4 months after prednisone were stopped). Clinical presentation resembled the previous ones and ANCA (proteinase 3) was again positive. We administered cyclophosphamide 150 mg/day for 3.5 months and prednisone 60 mg/day in a dose gradually decreasing to 10 mg/day until the third relapse appeared in November 2000 (41 months after the second relapse).
Renal affection was only of a moderate degree, with mild proteinuria and hematuria. Serum creatinine level increased from 129 to 181 μmol/L. Lung symptomatology deteriorated gradually with dyspnea, bloody sputum, and numerous bilateral lung granulomas on chest X-ray. Percutaneous lung biopsy (in April 2001) confirmed florid vasculitis with giant-cell granulomas. We administered two pulses of 1 g i.v. cyclophosphamide in a month interval and 20 mg of prednisone daily without any improvement in the patient status during the following 8 weeks. At that time, we noted a transient edema of the right upper limb, which progressed to vena cava superior syndrome in the next few months. Doppler sonography showed the patent right subclavian vein. With regard to the high cumulative alkylating agent’s dose, we started treatment with cyclosporin A 5 mg/kg/day (trough level 200–250 ng/mL) together with prednisone 20 mg every other day till October 2001. In these months, the patient’s condition continued to deteriorate with cough, bloody and purulent sputum and dyspnea, and with progressing vena cava superior syndrome. A chest X-ray and CT scan showed multiple nodular lung infiltrates up to 6 cm in diameter. Serum creatinine level increased temporarily to 398 μmol/L and creatinine clearance decreased to 0.302 mL/s. Therefore, we stopped cyclosporin administration and started chlorambucil in a dose of 8 mg/24 h.
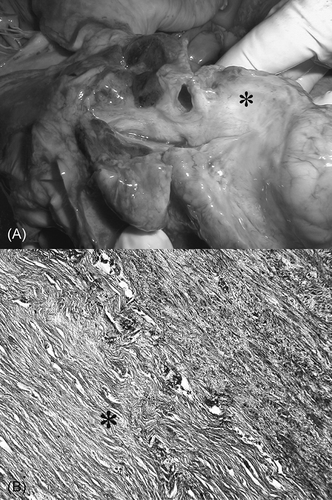
Figure 1. (A) The section through the left atrium with masses of fibrous tissue (asterisk), which compressed the large veins. (B) Almost acellular masses of the fibrous tissue 1.2 cm thick (asterisk) surrounding the lamina muscularis of the vena cava superior.
In spite of immunosuppressive treatment, dyspnea, cough, and hemorrhagic expectoration progressed and the chest X-ray displayed the increase in quantity and size of the granulomas. Vena cava superior syndrome deteriorated exhibiting a swollen face, neck, and upper right limb, with conspicuous superficial veins distension in these regions. Petechias and purpura appeared on the upper and lower limbs and on the soles. Herpetic and hemorrhagic efflorescence developed on buccal mucosa and the palate. A skin biopsy of these lesions resembled the purulent vasculitis and Osler’s noduli, but blood cultures were negative and echocardiography (and later autopsy) did not demonstrate vegetations on the heart valves. Serum creatinine level was 257 μmol/L, C-reactive protein was 164 mg/L. Anemia with hemoglobin 85 g/L progressed and the platelet count gradually decreased to 35 × 109/L. With regard to the patient’s heavy condition, chlorambucil was stopped. We continued in intravenous methylprednisolone, antimicrobial, antiviral, and supportive therapy. Due to hypoxia status, we started the mechanical ventilation. Treatment of renal failure by peritoneal dialysis was started. Patient died of respiratory failure in January 2002.
The autopsy confirmed the diagnosis of Wegener’s granulomatosis. Yellowish noduli up to 4 cm of diameters were found in the lungs. The kidneys were shrunken with numerous sclerotized glomeruli, and severe tubular atrophy and interstitial sclerosis were documented. Rigid, whitish, 12 mm thick tissue spread upward along the large vessels from their origin in the heart (A), and embedded ascending aorta, vena cava superior, and pulmonary artery. Vena cava superior was heavily constricted by masses of this tissue. In light microscopy, this tissue was composed of fibrous material without inflammatory cellulization (B).
DISCUSSION
The time course of this disease displays two important features:
| (1) | Sensitivity of the disease to alkylating agents decreased with the number of relapses. While clinical symptoms and laboratory signs of the first Wegener’s granulomatosis presentation vanished during 6 weeks of cyclophosphamide administration, it took 3.5 months course of daily cyclophosphamide and 1 year of alkylating agents administration to reach a partial remission lasting 3 years after the first relapse, and, in spite of aggressive immunosuppressive treatment, we were not able to suppress the activity and fatal progression during the last relapse of the disease. | ||||
| (2) | The duration of disease remission was directly related to the intensity and time span of immunosuppressive treatment with alkylating agents. The first partial remission lasted 1.5 years after a 6-week course of cyclophosphamide, while the second one persisted for 3 years after a 1-year course of alkylating agents (cyclophosphamide followed by chlorambucil), with comparable dose of prednisone in both periods. Twelve years from the beginning, the fourth episode of the disease was resistant to the intensive immunosuppressive, antibacterial, and supportive treatment and the patient died of respiratory failure caused by Wegener’s lesion of the lungs. | ||||
Many studies underlined the importance of long-lasting immunosuppression by alkylating agents followed by antimetabolites in Wegener’s granulomatosis.Citation1
Edema of the right limb was diagnosed as the vena cava superior compression. It was later confirmed by autopsy, but the origin of masses of fibrous tissue surrounding the large vessels remained unclear. This tissue did not display any signs of granulomatous vasculitis or other inflammatory activity as it was previously described in parajugular,Citation2 prevertebral,Citation3 or in abdominal periaortic tissue in patients with ANCA proteinase-3 positive Wegener’s granulomatosis.Citation4 The tissue, which embedded the vena cava superior, resembled that described repeatedly as fibrosing mediastinitis.Citation5 Such tissue was found in some patients with Histoplasma capsulatum infection, or rarely with tuberculosis, fungal infection, in patients with autoimmune diseases like Behçet disease, rheumatic fever, and Hodgkin disease. It has also been described in patients with other idiopathic diseases, like retroperitoneal fibrosis, sclerosing cholangitis, and Riedel thyroiditis, but it may also be idiopathic.Citation5 The mechanism of this fibrous mass formation remains unclear and its links to infectious and noninfectious or immunologic causes remain speculative. The histopathologic features strongly suggest its genesis from a chronic inflammatory process. Generally, any chronic inflammatory process, including vasculitides, may induce fibrosis.Citation6 The tissue may represent an abnormal immunopathologic reaction to still unknown antigen present in our patient with Wegener’s granulomatosis.
Thus, we demonstrate an unusual course of Wegener’s granulomatosis with fibrous tissue formation surrounding the large vessels upward from their origin in the heart that caused vena cava superior syndrome. Our patient serves to emphasize the heterogeneity of associations between fibrosing mediastinitis and inflammation.
To our knowledge such a case was not reported previously.
ACKNOWLEDGMENT
This study was supported by the Main Research Project No MSM 0021620819 (Czech Republic).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Jayne DRW. Update on the European vasculitis study group trials (EUVAS). Curr Opin Rheumatol. 2001;13:48–55.
- Hadden RD, Meikle D, Coulthard A, Gholkar A, Crawford PJ, Jackson MJ. Wegener’s granulomatosis presenting with bulbar palsy and bilateral jugular vein compression. Neurology. 1998;50:1923–1924.
- Barreto P, Pagnoux C, Luca L, . Dorsal prevertebral lesion in Wegener granulomatosis: Report on four cases. Joint Bone Spine. 2011; 78:88–91.
- Blockmans D, Baeyens H, Van Loon R, Lauwers G, Bobbaers H. Periaortitis and aortic dissection due to Wegener’s granulomatosis. Clin Rheumatol. 2000;19:161–164.
- Rossi SE, McAdams HP, Rosado-de-Christenson ML, Franks TJ, Galvin RJ. Fibrosing mediastinitis. Radiographics. 2001;21:737–757.
- Dunn EJ, Ulicny Jr., KS, Wright CB, Gottesman L. Surgical implications of sclerosing mediastinitis. A report of six cases and review of the literature. Chest. 1990;97:338–346.