Abstract
Augmenter of liver regeneration (ALR), the expression of which increased in rat kidneys after renal ischemia/reperfusion (I/R) injury, enhances renal tubular cell regeneration in vivo and in vitro. We aimed to investigate the effects of ALR on apoptosis of renal tubular cells after renal I/R injury in vivo and consider the possible mechanisms. Rats that were subjected to bilateral renal ischemia for 60 min followed by reperfusion were administered with either vehicle or recombinant human ALR (rhALR). Renal dysfunction and histologic injury were assessed by the measurement of serum biochemical markers and histological grading. Apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL). Caspase-3 activity was measured using a colorimetric protease assay. Expression of Bcl-2, Bax Fas, phosphorylated-Akt (p-Akt), and phosphorylated-p53 (p-p53) was determined by western blotting. Compared with vehicle-treated rats, renal dysfunction and histologic injury were significantly attenuated by administration of rhALR. The number of TUNEL-positive tubular cells and caspase-3 activity were decreased, Bcl-2 and p-Akt expression was up-regulated, and Bax and p-p53 expression was down-regulated by administration of rhALR. However, administration of rhALR had no effect on Fas protein expression. These results indicate that the protective effect of rhALR on renal I/R injury is associated with its anti-apoptotic action in renal tubular cells. RhALR inhibits apoptosis by increasing the ratio of Bcl-2 to Bax and by decreasing the activity of caspase-3. The activation of Akt and inactivation of p53 are involved in the rhALR anti-apoptosis process.
INTRODUCTION
Acute kidney injury (AKI) contributes significantly to the morbidity and mortality of hospitalized patients, despite significant advances in supportive care.Citation1,2 AKI is often multifactorial, but renal ischemia/reperfusion (I/R) is one of the major causes. Its underlying pathophysiological mechanism is complex, and although the loss of functional tubular epithelial cells is a major contributing factor to renal dysfunction in AKI, massive necrosis of tubular cells has only been found in a few segments in human biopsy specimens and experimental animal models.Citation3,4 This suggests that other factors, in addition to necrosis, may contribute to renal dysfunction in AKI.
Apoptosis has frequently been observed in animal models of kidney I/R injury and in human AKI,Citation5–7 and is thought to represent an important mechanism of renal dysfunction in AKI.Citation8 Significant progress in understanding the role of apoptosis and its triggering factors has been made in the last decade. Many growth factors (such as hepatocyte growth factor, α-melanocyte-stimulating hormone, erythropoietin and colony-stimulating-factor-1)Citation9–13 and drugs (such as glutamine and furosemide)Citation14,15 have been confirmed to attenuate AKI by inhibiting the apoptosis of renal tubular cells. In contrast, bortezomib and rapamycin aggravated AKI by enhancing tubular cell apoptosis.Citation16,17 Further investigations into the mechanisms responsible for tubular cell apoptosis and identification of the cytokines involved in the process may have important therapeutic implications.Citation18
Augmenter of liver regeneration (ALR) was purified from the liver of newborn rats and cloned by Hagiya et al.Citation19 Several studies have found that ALR can stimulate hepatocyte proliferation and protect the liver from acute liver failure.Citation20–22 Recent studies have shown that depletion of ALR in hepatocytes by anti-sense oligonucleotide transfection induced hepatocyte apoptosis, while recombinant human ALR (rhALR) significantly decreased hepatocyte apoptosis in vitro.Citation23,24 The expression of exogenous ALR increased the survival of hepatoma cells, accompanied by a decrease in apoptosis.Citation25 A protective effect of ALR on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells has also been found.Citation26 Overall, these findings suggest that ALR exhibits anti-apoptotic activity. ALR expression has been detected not only in the liver but also in the kidney, by Northern blotting.Citation19 We have previously shown that renal ALR expression significantly increased in rats with ischemic AKI and gentamicin-induced AKI, and that ALR could protect against AKI by enhancement of renal tubular cell regeneration.Citation27,28 We also confirmed that recombinant rat ALR (rrALR) could inhibit renal tubular cell apoptosis induced by gentamicin in vitro.Citation29 However, the ability of ALR to inhibit tubular cell apoptosis in ischemic AKI in vivo, and the possible mechanisms involved, remains unclear, and no relevant studies have been identified in the literature.
This study examined the effects of rhALR on renal functional impairment and histological damage by injection of the exogenous protein into AKI rats. To clarify the effects of rhALR on renal tubular cell apoptosis, as well as its possible mechanisms, we examined apoptosis using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) method, evaluated caspase-3 activity using a colorimetric protease assay, and determined Bcl-2, Bax, Fas, p-Akt, and p-p53 expression by western blotting in a rat model of ischemic AKI.
MATERIALS AND METHODS
Animals
Male Sprague–Dawley rats weighing 200–250 g were purchased from the Experimental Animal Center of Chongqing Medical University (Chongqing, China). Rats received a standard diet and water ad libitum and were housed under a 12 h light/dark cycle. All experimental procedures conformed to institutional guidelines for the care and use of laboratory animals in Chongqing Medical University, Chongqing, China, and to the National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996).
Study Design
Rats were randomized into four groups as follows: (1) sham-operated group; (2) I/R+rhALR1 group; (3) I/R+rhALR2 group; and (4) I/R+vehicle group. Rats were injected intraperitoneally with rhALR1 (100 μg/kg), rhALR2 (200 μg/kg), or vehicle (saline) at the end of ischemia and at 12, 24, 36, 48, and 60 h after reperfusion. Equal volumes of vehicle and rhALR (Institute for Viral Hepatitis, Chongqing Medical University, Chongqing, China, which we described previously)Citation30 were injected with respect to body weight, and they were administered at the same time points. Rats were killed at 24 and 72 h after reperfusion (eight rats per group for each time point).
Rat Model of Renal I/R
AKI rats were anesthetized with intraperitoneal (i.p.) sodium pentobarbital (50 mg/kg body weight). The core body temperature was maintained at 37°C during surgery using a homeothermic table. The abdominal cavity was exposed via a midline incision. Both renal pedicles were identified and occluded for 60 min with microvascular clamps. After removal of the renal clamps, the kidneys were observed for a further 5 min to ensure that they changed color, indicating blood reperfusion. Rats with incomplete reperfusion, judged visually, were not included in the statistical analyses. To compensate for the fluid loss during surgery, 1 mL saline/100 g body weight at 37°C was injected into the abdomen. The midline incision was closed after reperfusion. Sham-operated rats were subjected to an identical surgical procedure, without occlusion of the renal pedicles.
Biochemical Analysis
Blood samples were drawn by intracardiac puncture. The samples were centrifuged at 3000 rpm (1000 × g) for 15 min to separate serum. Serum samples were used for the measurement of renal biochemical parameters. Blood urea nitrogen and serum creatinine levels were evaluated using a Hitachi 747 automatic analyzer (Hitachi Co. Ltd., Tokyo, Japan).
Histological Examination
For histological examination, the harvested kidney from each animal was fixed immediately in 10% neutral buffered formalin solution. The kidney tissue block was dehydrated through a graded alcohol series, embedded in paraffin, cut into 4-μm sections, and then stained with hematoxylin and eosin. Histologic changes were evaluated by semiquantitative measurements of tissue damage. Tubular injury in the outer medulla was examined in 20 randomly selected ×200-field sections. Each tubular profile was assigned to one of five categories, according to the following criteria: 0, normal; 1, areas of tubular epithelial cell swelling, vacuolar degeneration, necrosis, and desquamation involving <25% of the tubular profile; 2, similar changes involving ≥25% but <50% of the tubular profile; 3, similar changes involving ≥50% but <75% of the tubular profile; and 4, similar changes involving ≥75% of the tubular profile. To minimize observer bias, the morphometric examination was performed in a blinded manner by two independent pathologists. The final score per section corresponded to the average histological damage score from both observers.
Detection of Apoptotic Cells
Sections were processed for TUNEL staining using an in situ apoptosis detection kit (R&D Co. Ltd., Minneapolis, MN, USA). Briefly, paraffin-embedded sections were deparaffinized in xylene for 5 min and rehydrated through graded concentrations of ethanol. After washing twice with phosphate-buffered saline (PBS) for 5 min, the sections were treated with 1.0 μg/mL proteinase K in PBS at 37°C for 15 min and washed with deionized water for 10 min. The tissue sections were incubated in 2% H2O2 at 37°C for 15 min and then rinsed with deionized water for 10 min to inactivate endogenous peroxidase. The slides were then incubated with a TdT buffer (25 mmol/L Tris-HCl buffer, pH 6.6, 0.2 mol/L potassium cacodylate, and 0.25 mg/mL bovine serum albumin) at room temperature for 30 min, and reacted with 0.1 U/μL TdT dissolved in a TdT buffer supplemented with 1.0 nmol/L digoxigenin (Dig)-dUTP in a humid chamber at 37°C for 1 h. Signals were detected immunohistochemically using a horseradish peroxidase (HRP)-conjugated sheep anti-Dig antibody. Quantitative measurement of apoptotic cells was achieved by examining 10 randomly selected ×200 fields in the outer medulla. To minimize observer bias, counting of the mean number of apoptotic cells per field was performed in a blinded manner by two independent pathologists.
Caspase-3 Activity Assay
Caspase-3 activity was determined using a commercial caspase-3 activity kit (Beyotime Institute of Biotechnology, Jiangsu, China). In brief, the renal cortices were homogenized in lysis buffer, and the lysate was centrifuged at 20,000 × g for 10 min at 4°C. Assays were performed on 96-well microtiter plates by incubating 10 μL supernatant in 80 μL reaction buffer (1% NP-40, 20 mol/L Tris-HCl (PH 7.5), 137 mmol/L NAD, and 10% glycerol) containing 10 μL caspase-3 substrate (Ac-DEVD-pNA) (2 mmol/L). Plates were incubated at 37°C for 1 h. Samples were measured using an enzyme-linked immunosorbent assay reader at an absorbance of 405 nm. Details of the analytical procedure are described in the manufacturer’s protocol. Relative caspase-3 activity was calculated as the ratio of the treated-group result to the sham-operated-group result.
Western Blot Analysis
Rat kidney tissue was minced in the presence of 50 mmol/L Tris, 0.1% NP-40, 0.15 mol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 5 mmol/L benzamidine, 1 mmol/L phenlymethylsulfonylfouride (PMSF), 1 μmol/L pepstatin, 1 μmol/L leupeptin, and 1 μmol/L aprotinin. The resultant pellet was homogenized and the supernatants were collected after centrifugation at 13,000 × g at 4°C for 20 min. Protein concentration was assessed using the Bradford Assay Method (Sigma Chemical Co., St. Louis, MO, USA). Aliquots of each sample containing 25 μg of protein were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After incubation in a blocking solution [5% nonfat dry milk in PBST (0.05% Tween 20 in PBS)] at room temperature for 1 h, the membrane was subjected to repeated washing in PBST (4 × 10 min) and then incubated with primary polyclonal antibodies in the blocking solution at 4°C overnight [1:200 rat anti-Bcl-2, Bax, Fas, p-Akt (Ser 473)/Akt (total Akt), and p-p53/p53(total p53), Santa Cruz Biotechnology, Santa Cruz, CA, USA]. After washing with PBST, the membrane was incubated for 1 h with HRP-tagged goat anti-rabbit IgG (Sigma Chemical Co.). Proteins were visualized by chemiluminescence. β-Actin was used as an internal control for each membrane.
Statistical Analysis
All data were expressed as mean ± standard deviation. Statistical analysis was carried out using SPSS 11.0 software. One-way analysis of variance was performed followed by the Student–Newman–Keuls test for parametric values or the Kruskal–Wallis–Dunn test for nonparametric values. p-Values <0.05 were considered statistically significant.
RESULTS
Effects of rhALR on Renal Dysfunction caused by I/R
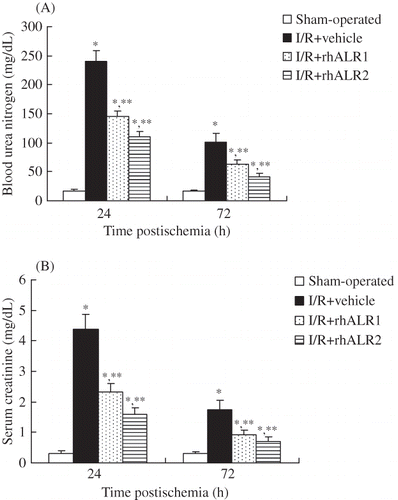
Rats subjected to renal I/R exhibited elevated blood urea nitrogen and serum creatinine levels compared with sham-operated rats (p < 0.05). Their peak levels at 24 h and lower levels at 72 h indicated that renal dysfunction reached peak at 24 h and began to restore thereafter. Compared with vehicle-treated animals, blood urea nitrogen and serum creatinine levels were significantly lowered in rats treated with rhALR1 or rhALR2 at 24 and 72 h, respectively (p < 0.05). Their levels in the I/R+ALR2 group were lower than those in the I/R+ALR1 group at both time points (p < 0.05) (A and B).
Figure 1. Effects of rhALR treatment on renal function. Rats subjected to renal I/R exhibited elevated blood urea nitrogen (A) and serum creatinine (B) levels compared with sham-operated rats. These elevated levels were significantly lowered in rats treated with rhALR1 or rhALR2 (p < 0.05). The levels of blood urea nitrogen and serum creatinine were lower in I/R+ALR2 rats than in I/R+ALR1 rats (p < 0.05).
Notes: Data shown are mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
Effects of rhALR on Histologic Alterations caused by Renal I/R
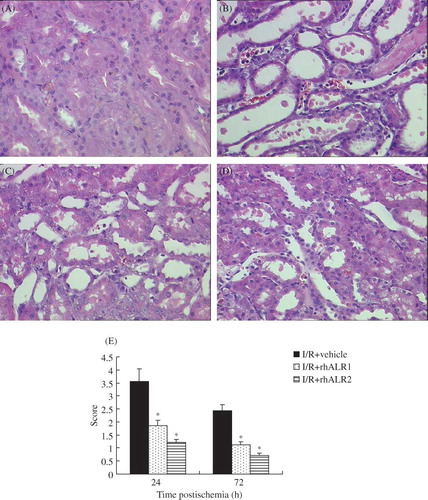
Renal I/R caused marked alterations in renal histology (B) compared with kidneys from sham-operated animals (A). Renal I/R caused widespread degeneration of tubular architecture, tubular dilation, swelling and necrosis, luminal congestion with loss of brush borders, and infiltration of polymorphonuclear neutrophils. The characteristic histological features of ischemic injury were evident at 24 h and were milder at 72 h after reperfusion. In contrast, renal sections obtained from animals treated with rhALR1 or rhALR2 demonstrated marked reduction of the histological characteristics of renal injury (C and D). Semiquantitative assessment of the histological lesions showed a significantly higher score in vehicle-treated rats compared with rhALR1- and rhALR2-treated rats after reperfusion. However, the scores in the rhALR2-treated rats were lower than those in the rhALR1-treated rats (p < 0.05, E).
Figure 2. Effects of rhALR on I/R-induced renal injury. Representative hematoxylin-eosin-stained renal sections from sham-operated (A) and vehicle- (B), rhALR1- (C), and rhALR2-treated (D) rats at 24 h, demonstrating more severe lesions of tubular necrosis in I/R rats. RhALR1 and rhALR2 treatment significantly preserved renal tissue morphology. Magnification ×400. Semiquantitative assessment of histological lesions based on tubular necrosis.
Notes: Values represent mean scores ± SD. *Denotes p < 0.05 versus the I/R+vehicle group.
Effects of rhALR on Tubular Apoptosis
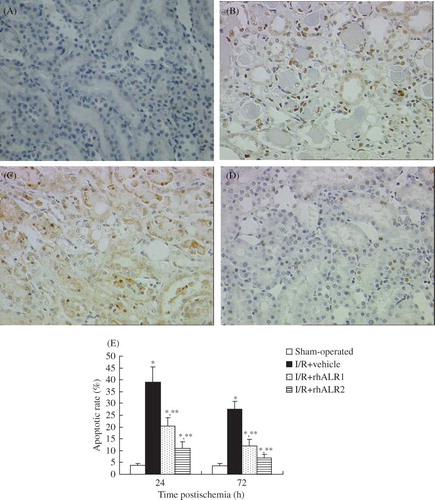
Tubular epithelial cell apoptosis was quantified in renal sections and confirmed by TUNEL staining. TUNEL-positive apoptotic cells were observed at 24 and 72 h after reperfusion. There were few apoptotic cells in the renal cortex or the outer stripe of the outer medulla in sham-operated rats (A). Kidneys from the animals subjected to renal I/R exhibited a marked increase in the number of TUNEL-positive cells, peaking at 24 h after reperfusion (B). The mean numbers of apoptotic cells significantly decreased in the I/R+rhALR1 (C) and I/R+rhALR2 (D) groups compared with the I/R+vehicle group at 24 and 72 h, respectively (p < 0.05). The apoptotic rate in the AKI+rhALR2 group was lower than that in the AKI+rhALR1 group at both time points (p < 0.05, E).
Figure 3. Effects of rhALR treatment on I/R-induced tubular cell apoptosis. Representative sections at 24 h are shown in (A–D). Apoptosis was evaluated by TUNEL staining. Few TUNEL-positive apoptotic cells were noted in the tubular lumen in the normal group (A), but increased numbers of TUNEL-positive cells were observed in the I/R group (B). Few TUNEL-positive cells were seen in the rhALR-treated group, indicating reduced apoptosis (C, D). Magnification ×200. (E) Mean number of apoptotic cells.
Notes: Data are expressed as mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
Effects of rhALR on caspase-3 Activity
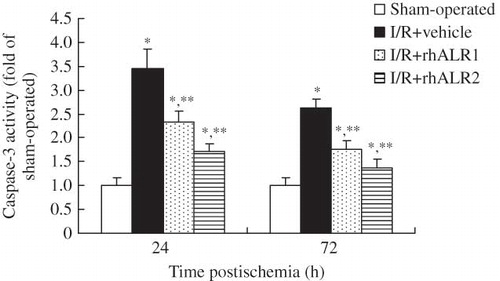
Caspase-3 activity significantly increased at 24 h in kidneys subjected to renal I/R and slightly increased at 72 h after reperfusion compared with sham-operated animals (p < 0.05). The elevated caspase-3 activity was significantly reduced by rhALR1 or rhALR2 administration compared with vehicle administration. Caspase-3 activity was lower in the I/R+rhALR2 group than in the I/R+ rhALR1 group at both time points (p < 0.05, ).
Figure 4. Effects of rhALR on caspase-3 activity. Caspase-3 activity significantly increased in kidneys subjected to renal I/R at 24 and 72 h compared with sham-operated animals. Elevated caspase-3 activity was significantly reduced by rhALR1 or rhALR2 administration compared with vehicle administration. Caspase-3 activity was lower in I/R+rhALR2 rats than in I/R+rhALR1 rats.
Notes: Data are expressed as mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
Effects of rhALR on Bcl-2 and Bax Protein Expression
Levels of Bcl-2 and Bax expression in kidneys from sham-operated rats were low. Expression levels of Bcl-2 and Bax increased after renal I/R, but the Bcl-2/Bax ratio significantly decreased at 24 and 72 h after reperfusion (p < 0.05). Up-regulation of Bcl-2 and down-regulation of Bax were observed in kidneys from rhALR-treated rats compared with vehicle-treated rats. The Bcl-2/Bax ratio was higher in the I/R+rhALR2 group than in the I/R+rhALR1 group at both time points (A–E).
Figure 5. Effects of rhALR treatment on Bcl-2 and Bax expression in kidneys of rats subjected to I/R injury. The expression levels of Bcl-2 (A, B) and Bax (C, D) were low in sham-operated rats, but increased in I/R+vehicle rats. Bcl-2 was up-regulated and Bax was down-regulated in I/R+rhALR1 and I/R+rhALR2 rats compared with I/R+vehicle rats. The Bcl-2/Bax protein ratio significantly increased in both rhALR-treated groups compared with vehicle-treated rats. The Bcl-2/Bax ratio was higher in I/R+rhALR2 rats than in I/R+rhALR1 rats (E).
Notes: Data are expressed as mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
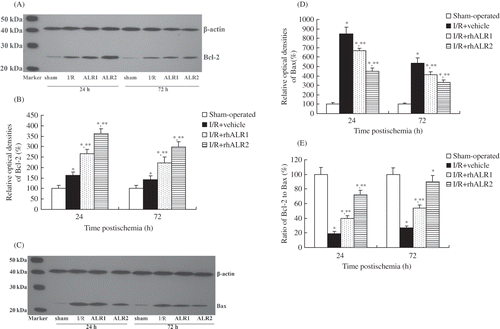
Effects of rhALR on p-Akt and p-p53 Protein Expression
Levels of p-Akt and p-p53 expression in kidneys from sham-operated rats were low. However, P-Akt expression decreased and p-p53 expression increased (p < 0.05) in the I/R+vehicle group at 24 h. The level of p-Akt was significantly higher in rhALR-treatment groups than in the I/R+vehicle group. The level of p-Akt was higher in the I/R+rhALR2 group than in the I/R+rhALR1 group. The level of p-p53 was significantly lower in rhALR-treatment groups than in the I/R+vehicle group. The level of p-p53 was lower in the I/R+rhALR2 group than in the I/R+rhALR1 group. There was no significant difference in the total Akt and total p53 expression among the four groups (A–F).
Figure 6. Effects of rhALR treatment on p-Akt and p-p53 expression in kidneys of rats subjected to I/R injury at 24 h. The expression levels of p-Akt (A, C) and p-p53 (D, F) were low in sham-operated rats, but p-Akt decreased and p-p53 increased in I/R+vehicle rats. p-Akt was up-regulated and p-p53 was down-regulated in I/R+rhALR1 and I/R +rhALR2 rats, as compared with I/R+vehicle rats. There was no significant difference in the total Akt (B) and total p53 (E) expression among the four groups.
Notes: Data are expressed as mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
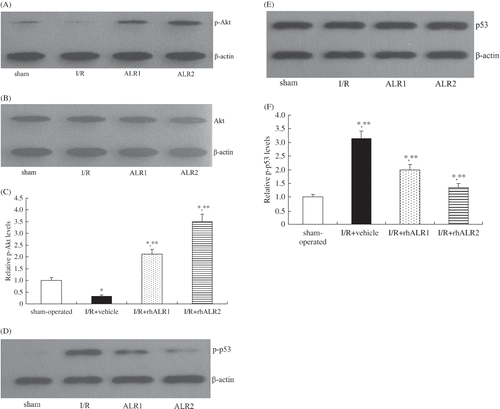
Effects of rhALR on Fas Protein Expression
Fas protein expression was detectable at low levels in sham-operated rats. It significantly increased in the I/R+vehicle group at 24 h and slightly increased at 72 h after reperfusion. RhALR1 or rhALR2 administration had no effect on Fas protein expression either 24 h or 72 h after reperfusion (A and B).
Figure 7. Effects of rhALR treatment on Fas expression in kidneys of rats subjected to I/R. Fas protein expression was detectable at low levels in sham-operated rats. It increased in I/R+vehicle rats at 24 h, and decreased at 72 h after reperfusion. RhALR1 and rhALR2 administration had no effect on Fas protein expression at either 24 or 72 h after reperfusion.
Notes: Data are expressed as mean ± SD.
*Denotes p < 0.05 versus the sham-operated group. **Denotes p < 0.05 versus the I/R+vehicle group.
DISCUSSION
Apoptosis is a physiological process involved in nephrogenesis and the maintenance of tissue homeostasis in the kidney. However, excessive apoptosis of renal tubular cells leads to tissue loss and renal dysfunction.Citation4 In this study, apoptosis of renal tubular cells and renal dysfunction peaked at 24 h after reperfusion, while fewer apoptotic cells, accompanying renal function repair, were detected at 72 h after reperfusion, suggesting that apoptosis within the first 72 h after renal I/R injury is one of the main causes for renal dysfunction in ischemic AKI. Our previous study showed that ALR expression in the kidney began to increase from 6 h and reached a peak at 72 h after renal I/R injury,Citation28 indicating that ALR may participate in the tubular cell apoptotic process in vivo. In this study, apoptosis rate of renal tubular cells decreased significantly in AKI rats treated with rhALR in a dose-dependent manner at both time points, accompanied by a marked amelioration of renal dysfunction and histological impairment, indicating that inhibiting renal tubular cell apoptosis is one of the main mechanisms of rhALR protecting against ischemic AKI.
ALR is a growth factor with mitogenic effects in hepatocytes, hepatocarcinoma cells, and renal tubular cells.Citation27–31 ALR belongs to a novel group of the so-called cytozymes on account of its action as a growth factor and a sulfhydryl oxidase, which binds FAD containing a redox-active CxxC disulfide proximal to a flavin ring.Citation32 Previous studies have shown that ALR activates the ras/MEK/ERK and PI3K/Akt pathways.Citation33 However, the influence of ALR on signaling pathways differs from that of other growth factors such as epidermal growth factor (EGF). ALR causes a transient increase in ERK phosphorylation compared with EGF’s permanent effect.Citation33 The PI3K/AKT and ERK pathways are also known to be important pathways for apoptosis. Although recent studies have reported anti-apoptotic effects of ALR in human hepatocytes, neuroblastoma cells, and hepatoma cells,Citation23–26 and rrALR has been shown to inhibit gentamicin-induced renal tubular cell apoptosis in vitro,Citation29 the mechanisms of ALR-inhibiting apoptosis remain unclear.
Caspase-3 is the most important executioner in most apoptotic signaling cascades. Significant evidence indicates that caspase-3 is either partially or totally responsible for the proteolytic cleavage of many key proteins, facilitates cellular disassembly, and serves as a marker of cells undergoing apoptosis.Citation34,35 In this study, caspase-3 activity increased significantly after renal I/R, reached a peak at 24 h, and increased slightly at 72 h. The elevated caspase-3 activity was significantly reduced by rhALR administration. The variation trend of caspase-3 activity was the same as that of apoptosis rate detected by TUNEL. These results again confirm that ALR can inhibit renal apoptosis in vivo and indicate that ALR inhibit renal apoptosis through inhibition of caspase-3 activity.
Upstream events of caspase-3 activation are thought to involve two main pathways: the mitochondrial and/or the death receptor pathways. Caspase-3 can be activated by caspase-9 in the mitochondrial pathway, which is in turn activated from procaspase-9 by cytosolic cytochrome c. The mitochondrial release of cytochrome c is regulated by Bcl-2 family proteins, including Bcl-2 and Bax, which bind to the mitochondrial outer membrane and block cytochrome c efflux. Bcl-2 and Bax have similar amino acid sequences but are functionally opposed: Bcl-2 inhibits apoptosis, whereas Bax counteracts this effect.Citation36–39 In this study, we found low levels of Bcl-2 and Bax expression in sham-operated rats, but these levels were increased by renal I/R, in accordance with previous research.Citation40 Administration of rhALR significantly increased Bcl-2 expression and decreased Bax expression, thus increasing the Bcl-2/Bax ratio. These results indicate that ALR inhibits apoptosis though the mitochondrial pathway.
Because the Bcl-2 family upstream regulator is identified to be Akt, also known as protein kinase (PK) B, a Ser-Thr kinase that plays an essential role in the regulation of cell survival, we speculated that changes in Bcl-2 and Bax expression due to rhALR may be mediated through activation of Akt kinase, which is believed to be mediated by phosphorylation of Akt at Ser 473.Citation41 To this end, we studied the effect of rhALR on Akt phosphorylation. As demonstrated in , treatment of I/R rats with rhALR caused dramatic activation of Akt, as shown by the rapid and marked phosphorylation of Akt protein at Ser 473. The activity of Akt is regulated by a variety of growth factors in a PI3-kinase-dependent mannerCitation42; for example, activation of PI3K/Akt by hepatocyte growth factor has been shown to deliver survival signals leading to inhibition of apoptosis in renal tubular cells.Citation43 Previous studies have shown that ALR activates the PI3K/Akt pathways in mediating hepatocyte proliferation.Citation33 These results suggest that rhALR may cause Bcl-2 and Bax expression changes by activation of the PI3K/Akt pathway.
As a transcription factor, the tumor suppressor gene p53 has been reported to induce the up-regulation of pro-apoptotic Bax and the down-regulation of anti-apoptotic Bcl-2. P53 may also have non-transcriptional actions inactivating Bcl-2/Bcl-xL and activating Bax.Citation44 P53 also boosts the activation of the caspase cascade by both transcription-dependent and transcription-independent mechanisms.Citation45 Furthermore, p53 activity is modulated by various mechanisms, and post-transcriptional phosphorylation and dephosphorylation may be the key regulatory steps. P53 is phosphorylated on numerous serines in both amino- and carboxy-terminal domains, and phosphorylation of p53 at Ser 15 leads to a reduced interaction between p53 and its negative regulator MDM2. MDM2 inhibits p53 accumulation by targeting it for ubiquitination and proteasomal degradation.Citation46 Our experiments demonstrated a drastic decrease in p53 phosphorylation in rhALR-administrated rats, which indicates that rhALR increases the Bcl-2/Bax ratio by inactivating p53.
Fas–FasL interactions in the death receptor pathway lead to processing of procaspase-8 to caspase-8; activated caspase-8 is the most upstream caspase in the Fas apoptotic pathway and promotes caspase-3 activation.Citation38,39,47 Fas protein expression in this study was detectable at low levels in the sham-operated group and was increased by I/R, indicating that the death receptor pathway participates in the process of tubular cell apoptosis. However, rhALR administration had no effect on Fas protein expression. Participation of the entire death pathway cannot be ruled out with the data presented. Other receptors or ligands of the death pathway should be detected in future studies.
The underlying pathophysiological mechanisms of ischemic AKI are complex. Apoptosis is one of the main causes; other causes such as proliferation and necrosis of tubular cells and renal inflammation also play an important role.Citation48 We have found that ALR could protect against ischemic AKI by reducing necrosis of tubular cells, enhancing renal tubular cell regeneration,Citation28 and inhibiting renal inflammation (data were not shown). In this study, we found that inhibiting renal tubular cell apoptosis was another mechanism by which ALR protects against ischemic AKI.
In conclusion, we investigated the effects of rhALR on I/R-induced apoptosis and the expression of apoptosis signal transduction genes in a rat model of surgical ischemic AKI. RhALR significantly attenuated I/R-induced renal dysfunction and histological impairment, including the apoptosis of renal tubular cells. These results suggest that the inhibition of I/R-induced apoptosis by rhALR is mediated by the intrinsic Bcl-2-mediated mitochondrial signaling pathway. The activation of Akt and inactivation of p53 are possibly involved in the rhALR anti-apoptosis process.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Natural Scientific Foundation of China (no. 39071364, 81000299) and the Natural Science Foundation Project of CQ CSTC (CSTC, 2010BB5385), and grants from the Applied Basic Research Programs of the Public Health Bureau Foundation of Chongqing Province (Nos. 2008-1-44 and 2010-2-126).
REFERENCES
- Ostermann M, Chang RW. Acute kidney injury in the intensive care unit according to RIFLE. Crit Care Med. 2007;35:1837–1843.
- Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am J Kidney Dis. 2009;53:961–973.
- Racusen LC, Fivush BA, Li YL, Slatnik I, Solez K. Dissociation of tubular cell detachment and tubular cell death in clinical and experimental “acute tubular necrosis”. Lab Invest. 1991;64: 546–556.
- Ueda N, Shah SV. Tubular cell damage in acute renal failure — apoptosis, necrosis, or both. Nephrol Dial Transplant. 2000;15:318–323.
- Safirstein RL. Acute renal failure: From renal physiology to the renal transcriptome. Kidney Int Suppl. 2004;91:S62–S66.
- Han KH, Kim HY, Croker BP, . Effects of ischemia-reperfusion injury on renal ammonia metabolism and the collecting duct. Am J Physiol Renal Physiol. 2007;293:F1342–F1354.
- Srichai MB, Hao C, Davis L, Golovin A, . Apoptosis of the thick ascending limb results in acute kidney injury. J Am Soc Nephrol. 2008;19:1538–1546.
- Bonegio R, Lieberthal W. Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens. 2002;11: 301–308.
- Dai C, Yang J, Liu Y. Single injection of naked plasmid encoding hepatocyte growth factor prevents cell death and ameliorates acute renal failure in mice. J Am Soc Nephrol. 2002;13:411–422.
- Jo SK, Yun SY, Chang KH, . Alpha-MSH decreases apoptosis in ischemic acute renal failure in rats: Possible mechanism of this beneficial effect. Nephrol Dial Transplant. 2001;16: 1583–1591.
- Vesey DA, Cheung C, Pat B, Endre Z, Gobé G, Johnson DW. Erythropoietin protects against ischemic acute renal injury. Nephrol Dial Transplant. 2004;19:348–355.
- Spandou E, Tsouchnikas I, Karkavelas G, . Erythropoietin attenuates renal injury in experimental acute renal failure ischemic/reperfusion model. Nephrol Dial Transplant. 2006;21:330–336.
- Menke J, Iwata Y, Rabacal WA, . CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. J Clin Invest. 2009;119:2330–2342.
- Kim YS, Jung MH, Choi MY, . Glutamine attenuates tubular cell apoptosis in acute kidney injury via inhibition of the c-Jun N-terminal kinase phosphorylation of 14-3-3. Crit Care Med. 2009;37:2033–2044.
- Aravindan N, Aravindan S, Riedel BJ, Wang HR, Shaw AD. Furosemide prevents apoptosis and associated gene expression in a rat model of surgical ischemic acute renal failure. Ren Fail. 2007;29:399–407.
- Huber JM, Tagwerker A, Heininger D, Mayer G, Rosenkranz AR, Eller K. The proteasome inhibitor bortezomib aggravates renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2009;297:F451–F460.
- Lieberthal W, Fuhro R, Andry CC, . Rapamycin impairs recovery from acute renal failure: Role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol. 2001;281:F693–F706.
- Rana A, Sathyanarayana P, Lieberthal W. Role of apoptosis of renal tubular cells in acute renal failure: Therapeutic implications. Apoptosis. 2001;6:83–102.
- Hagiya M, Francavilla A, Polimeno L, . Cloning and sequence analysis of the rat augmenter of liver regeneration (ALR) gene: Expression of biologically active recombinant ALR and demonstration of tissue distribution. Proc Natl Acad Sci USA. 1994;91:8142–8146.
- Zhang LM, Liu DW, Liu JB, . Effect of naked eukaryotic expression plasmid encoding rat augmenter of liver regeneration on acute hepatic injury and hepatic failure in rats. World J Gastroenterol. 2005;11:3680–3685.
- Tanigawa K, Sakaida I, Masuhara M, Hagiya M, Okita K. Augmenter of liver regeneration (ALR) may promote liver regeneration by reducing natural killer (NK) cell activity in human liver diseases. J Gastroenterol. 2000;35:112–119.
- Gao CF, Zhou FG, Wang H, Huang YF, Ji Q, Chen J. Genetic recombinant expression and characterization of human augmenter of liver regeneration. Dig Dis Sci. 2009;54:530–537.
- Thirunavukkarasu C, Wang LF, Harvey SA, . Augmenter of liver regeneration: An important intracellular survival factor for hepatocytes. J Hepatol. 2008;48:578–588.
- Ilowski M, Kleespies A, de Toni EN, . Augmenter of liver regeneration (ALR) protects human hepatocytes against apoptosis. Biochem Biophys Res Commun. 2011;404:148–152.
- Cao Y, Fu YL, Yu M, . Human augmenter of liver regeneration is important for hepatoma cell viability and resistance to radiation-induced oxidative stress. Free Radic Biol Med. 2009;47:1057–1066.
- Lorenzo P, Barbara P, Thomas L, . Protective effect of augmenter of liver regeneration on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic Res. 2009;43:865–875.
- Liao XH, Zhang L, Tang XP, Liu Q, Sun H. Expression of augmenter of liver regeneration in rats with gentamicin-induced acute renal failure and its protective effect on kidney. Ren Fail. 2009;31:946–955.
- Liao XH, Zhang L, Liu Q, Sun H, Peng CM, Guo H. Augmenter of liver regeneration protects kidneys from ischemia/reperfusion injury in rats. Nephrol Dial Transplant. 2010;25:2921–2929.
- Liao XH, Zhang L, Liu Q, . Effect of recombinant rat augmenter of liver regeneration on proliferation and apoptosis of renal tubular cells. Chin J Nephrol. 2005;21:747–751.
- Liu Q, Yu HF, Sun H, Ma HF. Expression of human augmenter of liver regeneration in Pichia pastoris yeast and its bioactivity in vitro. World J Gastroenterol. 2004;10:3188–3190.
- Pawlowski R, Jura J. ALR and liver regeneration. Mol Cell Biochem. 2006;288:159–169.
- Daithankar VN, Farrell SR, Thorpe C. Augmenter of liver regeneration: Substrate specificity of a flavin-dependent oxidoreductase from the mitochondrial intermembrane space. Biochemistry. 2009;48:4828–4837.
- Ilowski M, Putz C, Weiss TS, . Augmenter of liver regeneration causes different kinetics of ERK1/2 and Akt/PKB phosphorylation than EGF and induces hepatocyte proliferation in an EGF receptor independent and liver specific manner. Biochem Biophys Res Commun. 2010;394:915–920.
- Yang B, Elias JE, Bloxham M, Nicholson ML. Synthetic small interfering RNA down-regulates caspase-3 and affects apoptosis, IL-1 β, and viability of porcine proximal tubular cells. J Cell Biochem. 2011;112:1337–1347.
- Yang B, Hosgood SA, Nicholson ML. Naked small interfering RNA of caspase-3 in preservation solution and autologous blood perfusate protects isolated ischemic porcine kidneys. Transplantation. 2011;91:501–507.
- Shu D, Qing Y, Tong Q, . Deltonin isolated from Dioscorea zingiberensis inhibits cancer cell growth through inducing mitochondrial apoptosis and suppressing Akt and Mitogen activated protein kinase signals. Biol Pharm Bull. 2011;34:1231–1239.
- Chen NG, Lu CC, Lin YH, . Proteomic approaches to study epigallocatechin gallate-provoked apoptosis of TSGH-8301 human urinary bladder carcinoma cells: Roles of AKT and heat shock protein 27-modulated intrinsic apoptotic pathways. Oncol Rep. 2011;26:939–947.
- Alaimo A, Gorojod RM, Kotler ML. The extrinsic and intrinsic apoptotic pathways are involved in manganese toxicity in rat astrocytoma C6 cells. Neurochem Int. 2011;59:297–308.
- Zakeri Z, Lockshin RA. Cell death: History and future. Adv Exp Med Biol. 2008;615:1–11.
- Johnson DW, Pat B, Vesey DA, Guan Z, Endre Z, Gobe GC. Delayed administration of darbepoetin or erythropoietin protects against ischemic acute renal injury and failure. Kidney Int. 2006;69:1806–1813.
- Stiles BL. PI-3-K and AKT: Onto the mitochondria. Adv Drug Deliv Rev. 2009; 61(14):1276–1282.
- Franke T, Kaplan D, Cantley L. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997;88:435–437.
- Liu Y. Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol. 1999;277: F624–F633.
- Levine AJ, Hu W, The FZ. P53 pathway: What questions remain to be explored? Cell Death and Differentiation. 2006;13: 1027–1036.
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci. 2003;116:4077–4085.
- Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910–1915.
- Zhang H, Hile KL, Asanuma H, . IL-18 mediates proapoptotic signaling in renal tubular cells through a Fas ligand-dependent mechanism. Am J Physiol Renal Physiol. 2011;301:F171–F178.
- Lu CY, Hartono J, Senitko M, Chen JL. The inflammatory response to ischemic acute kidney injury: A result of the “right stuff” in the “wrong place”? Curr Opin Nephrol Hypertens. 2007;16:83–89.