Abstract
Objective: Renal fibrosis is a common cause of renal dysfunction with chronic kidney disease. We previously investigated the renoprotective effects of the antifibrotic agent pirfenidone in a rat model of subtotal nephrectomy. Here, we further evaluated the antifibrotic effects of pirfenidone in rat proximal tubular epithelial cells. Methods: NRK52E cells were incubated in a medium containing either transforming growth factor (TGF)-β1 (3 ng/mL) or platelet-derived growth factor (PDGF)-BB (5 Ang/mL) or both, with or without pirfenidone (0.1–1 mmol/L), for 24 h to assess mRNA expression, for 48 h to assess protein production, and for 1 h or various time (5–120 min) to assess phosphorylation of signal kinase. Results: TGF-β1, a key mediator in renal fibrosis, induced increases in the mRNA expression of various profibrotic factors and extracellular matrix, including plasminogen activator inhibitor type 1 (PAI-1), fibronectin, type 1 collagen, and connective tissue growth factor (CTGF)—increases which pirfenidone significantly inhibited. Specifically, pirfenidone potently inhibited TGF-β1-induced increases in the mRNA expression and protein secretion of PAI-1, an effect mediated, at least in part, via the mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling. Further, PDGF-BB, which has been implicated in renal interstitial fibrosis, potently activated PAI-1 expression under TGF-β1 stimulation, and pirfenidone significantly inhibited TGF-β1- and PDGF-BB-induced increases in PAI-1 expression. Conclusions: Taken together, these results suggest that TGF-β1 closely correlates with renal fibrosis in cooperation with several fibrosis-promoting molecules, such as PAI-1 and PDGF, in rat proximal tubular epithelial cells, and pirfenidone inhibits TGF-β1-induced fibrosis cascade and will therefore likely exert antifibrotic effects under pathological conditions.
INTRODUCTION
Chronic kidney disease is a critical problem in patients with diabetes, hypertension, and cardiovascular disease, and the prevalence of this disease has dramatically increased worldwide. Given that renal fibrosis is the final common pathway for nearly all forms of chronic kidney disease, and the progression of fibrosis coincides with the degree of renal dysfunction,Citation1,2 the treatment of renal fibrosis is believed to be a way of preventing the progression of chronic kidney disease.Citation3,4 A number of fibrotic diseases, including kidney, liver, and pulmonary fibrosis, are characterized by the accumulation of extracellular matrix (ECM) components.Citation5 Current evidence suggests that transforming growth factor (TGF)-β1 is largely responsible for this accumulation through its actions to induce production of ECM, to inhibit degradation, and to increase integrin expression, thereby enhancing ECM deposition.Citation6,7 In addition, TGF-β1 is known to induce the expression of additional profibrotic mediators, such as plasminogen activator inhibitor type 1 (PAI-1) and connective tissue growth factor (CTGF), which have been shown to play a role in the pathogenesis of renal fibrosis.Citation8,9 Moreover, TGF-β1 induces epithelial–mesenchymal transition (EMT), which is widely accepted as a mechanism by which injured renal epithelial cells transform into matrix-producing myofibroblasts that contribute to the development of fibrosis in chronic renal failure.Citation10–12 The important role of TGF-β1 in renal fibrosis is clearly demonstrated by the finding that renal scarring can be induced by overexpressing TGF-β1 but is inhibited by a neutralizing TGF-β1 antibody, decorin, or antisense in a number of animal models.Citation13 Given these findings thus far, TGF-β1 clearly plays a significant role in the progression of renal fibrosis.
Pirfenidone is an antifibrotic molecule that has been shown to prevent and even reverse ECM accumulation in experimental models of pulmonary fibrosis and has been launched for the treatment for idiopathic pulmonary fibrosis in Japan.Citation14,15 Pirfenidone has been shown to be effective in treating several fibrotic diseases, including not only pulmonary but also renal and liver fibrosis, as well as multiple sclerosis, conditions that share the pathology of abnormal ECM accumulation.Citation16 Pirfenidone was found to attenuate renal fibrosis in subtotal nephrectomized rats, a widely used model of progressive renal failure, and unilateral ureteral obstruction rats, a well-characterized model of tubulointestinal fibrosis.Citation17,18 We have also investigated the role of pirfenidone in renal function and histopathological changes in subtotal nephrectomized rats and demonstrated that pirfenidone alleviates the progression of renal failure via the decreases in the expression of TGF-β1 and ECM.Citation19 Although the mechanism underlying the antifibrotic action of pirfenidone remains poorly understood, the suggestion has been made that pirfenidone is associated with renal fibrosis in TGF-β1 expression.Citation20,21 In addition, pirfenidone also modulates the activities of additional mediators including growth factors, chemokines, and matrix metalloproteinases. Effects on growth factors include downregulation of platelet-derived growth factor (PDGF).Citation22 PDGF comprises a family of four isoforms (PDGF-A, B, C, and D), all of which are constitutively or inducibly expressed in renal cells, involved in the regulation of cell proliferation, migration, and accumulation of ECM, and implicated in the development of renal fibrosis.Citation23,24 In particular, the role of PDGF-B and D isoforms in mediating mesangioproliferative changes and development of glomerulosclerosis is well established.Citation25 In addition, PDGF-BB (homodimer) has an additive action on TGF-β1-induced EMT in renal epithelial cells.Citation26 Further, in the pathogenesis of renal interstitial fibrosis, different macrophage populations appear and produce both TGF-β1 and PDGF after renal injury as fibrogenic growth factors, which subsequently induce overproduction of ECM by myofibroblasts, leading to progressive renal fibrosis. Taken together, the above findings suggest that PDGF closely correlates with renal fibrosis induced by TGF-β1. However, no study has yet performed a detailed examination of its participation in renal fibrosis or the effects of pirfenidone against the factor. Here, we further evaluated the antifibrotic properties of pirfenidone against TGF-β1 and PDGF in cultured rat renal tubular epithelial cells.
MATERIALS AND METHODS
Reagents
Pirfenidone was synthesized at Astellas Pharma Inc. (Ibaraki, Japan). The mitogen-activated protein kinase (MEK) inhibitor U0126 was purchased from Biaffin GmbH & Co. KG (Kassel, Germany). NRK52E cells were purchased from American Type Culture Collection (Rockville, MD, USA). Recombinant human TGF-β1 and rat PDGF-BB were obtained from R&D Systems (Minneapolis, MN, USA). Primary detection antibodies used for Western blotting, phospho-MEK1/2, phospho-ERK1/2, phospho-c-Fos, phospho-SMAD2, phospho-p38 MAPK, phospho-c-Jun, and β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA) and detected using polyclonal anti-rabbit IgG conjugated to horseradish peroxidase (Medical & Biological Laboratories, Nagoya, Japan).
Cell Culture
NRK52E cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% fetal bovine serum (FBS) at 37°C in a humidified 95% air/5% CO2 atmosphere. For experiments, subconfluent cells in a 12-well plate were starved for 24 h by incubation with DMEM with 0.5% FBS and were then treated with TGF-β1 (3 ng/mL) and/or PDGF-BB (5 ng/mL) for 24 h to assess mRNA expression, for 48 h to assess protein production, and for 1 h or various time (5–120 min) to assess phosphorylation of kinases. When testing for inhibitory properties, pirfenidone or U0126 was added 30 min before treatment with either TGF-β1 or PDGF-BB or both. Samples were obtained at each time point and stored at −80°C until analysis by mRNA quantification, enzyme-linked immunosorbent assay (ELISA), or Western blotting.
mRNA Quantification
Culture medium was removed, and total RNA was extracted from the cell with the RNeasy Mini Kit (QIAGEN, Tokyo, Japan). Gene expression was assessed by real-time PCR using the TaqMan ABI 9700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The expression of target genes was normalized to the expression of β-actin mRNA for endogenous control. Sequences used as primers are as follows: PAI-1, forward 5′-GACAATGGAAGAGCAA CATG-3′, reverse 5′-ACCTCGATCTTGACCTTTT G-3′; fibronectin, forward 5′-CATGGCTTTAGGCG AACCAC-3′, reverse 5′-ATCTACATTCGGCAGGT ATGGTC-3′; type I collagen α1, forward 5′-CAACAA TTCCTGGCGTTACCTT-3′, reverse 5′-AAGCCCT GTATTCCGTCTCCTT-3′; CTGF, forward 5′-TCT TCGGTGGGTCCGTGT-3′, reverse 5′-TGGTATT TGCAACTGCTTTGGA-3′; α-smooth muscle actin (α-SMA), forward 5′-CGACATGGAAAAGATCTGG C-3′, reverse 5′-GGATCTTCATGAGGTAGTCG-3′; vimentin, forward 5′-GGGCGACCTCTACGAGG AG-3′, reverse 5′-TGTCATTGGTGAGCTGATC CA-3′; and β-actin, forward 5′-TCTGTGTGGATT GGTGGCTCTA-3′, reverse 5′-CATCGTACTCC TGCTTGCTGATC-3′.
PAI-1 Protein Measurement
Total PAI-1 protein amount in the cell culture supernatant was determined using a rat PAI-1 total antigen ELISA kit (Molecular Innovations, Novi, MI, USA) according to the manufacturer’s instructions.
Western Blotting
Cell culture supernatant was removed, and the cells were washed with phosphate buffered saline (PBS) and lysed in an ice-cold lysis buffer containing 137 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), 20 mM Tris-HCl (pH 7.9), 1% Triton-X100, and a protease inhibitor cocktail (Boeringer Mannhein, Mannheim, Germany). Cell lysates were diluted with Laemmli sample buffer (Bio-Rad Laboratories, Tokyo, Japan), resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed with the indicated antibodies, and immunoreactive bands were detected with chemiluminescence using Amersham ECL Plus (GE Healthcare, Wauwatosa, WI, USA). Detected bands versus β-actin bands were then quantified and normalized using ImageJ (National Institutes of Health, Washington DC, USA).
Statistical Analysis
Experimental results are expressed as mean ± standard error. Significant differences between pairs of groups were determined using Student’s t-test, whereas those between multiple groups were determined using Dunnett’s multiple range test. Significance was set at p < 0.05. Statistical and data analyses were conducted using the SAS 8.2 software package (SAS Institute Japan, Ltd., Tokyo, Japan).
RESULTS
Effect of Pirfenidone on TGF-β1-Induced mRNA Expression
In cultured NRK52E cells, TGF-β1 (3 ng/mL) significantly upregulated mRNA expression of PAI-1, ECM (fibronectin, type 1 collagen, and CTGF) and EMT markers (α-SMA and vimentin) (). Pirfenidone (0.1–1 mmol/L) concentration dependently and significantly suppressed TGF-β1-induced increases in PAI-1 and ECM expression, with PAI-1 expression being the most strongly inhibited of all (62% inhibition at 1 mmol/L pirfenidone), and exerted no significant effects on expression of EMT markers. In addition, TGF-β1 markedly increased levels of PAI-1 protein in the supernatant of extracellular signal-regulated kinase 52E (ERK52E) cells, whereas pirfenidone significantly suppressed this production (44% inhibition at 1 mmol/L; ).
Figure 1. Effect of pirfenidone on TGF-β1-induced mRNA expression of PAI-1 (A), fibronectin (B), type I collagen α1 (C), CTGF (D), α-SMA (E), and vimentin (F) in NRK52E cells. Cells were treated with TGF-β1 (3 ng/mL) for 24 h with or without pirfenidone (0.1–1 mmol/L). Values are mean ± standard error of four independent experiments. *p < 0.05 versus vehicle group; **p < 0.05 versus TGF-β1-treated group.
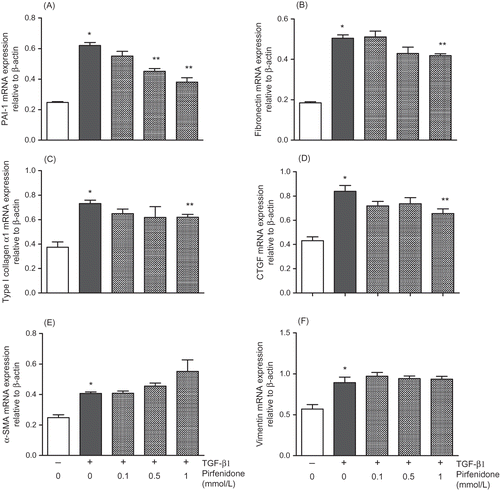
Figure 2. Effect of pirfenidone on TGF-β1-induced PAI-1 production in NRK52E cells. Cells were treated with TGF-β1 (3 ng/mL) for 48 h with or without pirfenidone (0.1–1 mmol/L). Values are mean ± standard error of four independent experiments. *p < 0.05 versus vehicle group; **p < 0.05 versus TGF-β1-treated group.
Effects of Pirfenidone on TGF-β1-Induced Phosphorylation of Signal Kinase
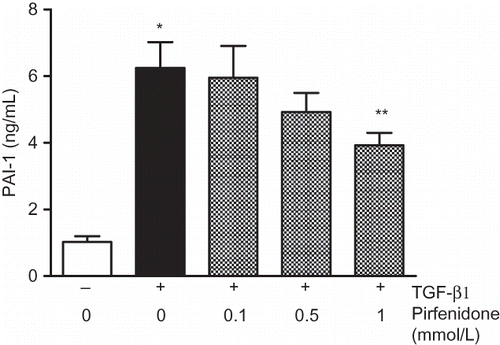
In Western blot studies, TGF-β1 increased phosphorylation of MEK1/2, ERK1/2, c-Fos, SMAD2 (A), and c-Jun (data not shown). In these signal transduction pathways, pirfenidone suppressed the phosphorylation of both ERK1/2 and c-Fos but exerted no effects on other pathways. B shows the phosphorylation time course of ERK1/2. Exposure to TGF-β1 enhanced ERK1/2 phosphorylation with a peak at 30 min, declining thereafter. The MEK inhibitor U0126 completely suppressed the TGF-β1-enhanced ERK1/2 phosphorylation to below basal levels over the study period. In contrast, pirfenidone only partially suppressed the increase in phosphorylation of ERK1/2 induced by TGF-β1. Calculation using imaging analysis showed that the inhibition rates of pirfenidone and U0126 at 30 min were 41% and 94%, respectively.
Figure 3. (A) Effect of pirfenidone on TGF-β1-induced phosphorylation of signal kinase in NRK52E cells. Cells were treated with TGF-β1 (3 ng/mL) for 1 h with or without pirfenidone. (B) Time-course effect of pirfenidone and U0126 on TGF-β1-induced phosphorylation of ERK1/2 in NRK52E cells. Cells were treated with TGF-β1 (3 ng/mL) for various durations (5–120 min) with or without pirfenidone (0.5 mmol/L) or U0126 (1 μmol/L). Western blot images were quantified densitometrically. N, nontreatment; T, TGF-β1; P, TGF-β1 + pirfenidone; U, TGF-β1 + U0126.
Effects of Pirfenidone on TGF-β1- and PDGF-BB-Induced mRNA Expression
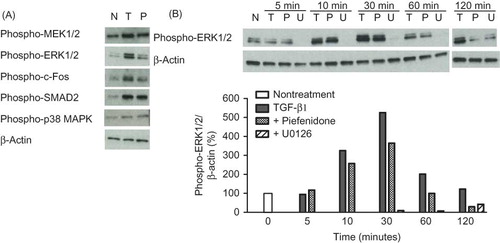
PDGF-BB (5 ng/mL) slightly increased the expression of PAI-1 and type I collagen but did not affect fibronectin, CTGF, or α-SMA levels (), and pirfenidone significantly suppressed these increases in PAI-1 expression. While PDGF-BB alone only weakly induced PAI-1 expression, the combined administration of TGF-β1 and PDGF-BB produced a synergistic increase in PAI-1 expression, which was subsequently potently suppressed by pirfenidone (A). Expression of type I collagen and CTGF were also slightly upregulated by co-treatment with TGF-β1 and PDGF-BB and similarly suppressed by pirfenidone (C and D).
Figure 4. Effect of pirfenidone on TGF-β1- and PDGF-BB-induced mRNA expression of PAI-1 (A), fibronectin (B), type I collagen α1 (C), CTGF (D), and α-SMA (E) in NRK52E cells. Cells were treated with either TGF-β1 (3 ng/mL) or PDGF-BB or both (5 ng/mL) for 24 h with or without pirfenidone (0.5 mmol/L). Values are mean ± standard error of four independent experiments. *p < 0.05 versus vehicle group; **p < 0.05 versus TGF-β1-treated group; ***p < 0.05 versus TGF-β1, PDGF-BB, or TGF-β1 + PDGF-BB-treated group.
DISCUSSION
Renal fibrosis represents a final common pathway associated with a variety of progressive kidney disorders, with pharmacological interventions that can reverse or prevent the progression of renal fibrosis being in high demand. As such, discovery of antifibrotic agents and investigation of effective mechanisms will greatly aid in ameliorating renal diseases. Recent studies have found pirfenidone to exhibit renoprotective activity in several animal models of diseases such as anti-Thy 1 antibody glomerulonephritis, progressive renal fibrosis with unilateral ureteral obstruction, and chronic renal failure in remnant kidney.Citation17,18,27 In these models, pirfenidone not only decreased ECM accumulation in the kidney but also prevented the progression of sclerotic glomerular and tubulointestinal lesions. These beneficial effects of pirfenidone seem to be mediated, at least partly, by decreasing the expression of TGF-β1. We also investigated the beneficial effects of pirfenidone on renal function and histopathological changes in 5/6 nephrectomized rats, with previous findings suggesting that pirfenidone exerts a renoprotective effect, reducing expression of TGF-β1 and ECM accumulation in the kidney.Citation19 Pirfenidone’s beneficial effects on renal fibrosis therefore make it an attractive option for the treatment of renal disorders. While TGF-β1 is known to play a role in a number of cellular processes, including fibrosis, relatively few TGF-β1 signaling pathway activations have been identified, and the mechanism underlying the antifibrotic action of pirfenidone behind the suggested TGF-β1 activation pathways remains poorly understood as well. We therefore evaluated the antifibrotic properties of pirfenidone against TGF-β1 and several profibrotic factors in cultured renal tubular epithelial cells.
We first investigated the inhibitory effect of pirfenidone on TGF-β1-induced increases in expression of ECM and several profibrotic factors. Pirfenidone significantly suppressed TGF-β1-induced increases in PAI-1 and ECM expression, with PAI-1 expression being the most strongly inhibited of all; pirfenidone also significantly inhibited TGF-β1-induced increases in PAI-1 protein which could be produced by both epithelial cells and transformed myofibroblasts.Citation28,29 Further, while TGF-β1 has been reported to markedly increased PAI-1 mRNA expression and protein in cultured glomeruli, these increases were inhibited by pirfenidone.Citation30 The glomerular mesangial and type IV collagen-positive area, renal collagen content, and PAI-1 mRNA levels were all significantly increased in the kidneys of mouse models of aldosterone/salt-induced renal fibrosis.Citation31 Given that PAI-1 is the major inhibitor of plasminogen activators, elevated levels of this factor are believed to inhibit plasmin generation and activation of matrix-degrading metalloproteinases, thereby decreasing ECM degradation and enhancing ECM accumulation in the kidney. In addition, TGF-β1 and PAI-1 mRNA expression has been found to be increased in rats with cyclosporine-induced nephrotoxicity; pirfenidone administration reduced expression of these factors and subsequently improved renal function.Citation32 Taken together, these data suggest that TGF-β1 modulates renal fibrosis, in part, through increasing levels of PAI-1, a major inhibitor of ECM degradation, with the antifibrotic effects of pirfenidone ostensibly mediated partly by reducing these elevated levels of PAI-1. These present and previous findings therefore suggest that TGF-β1-induced PAI-1 expression and subsequent ECM accumulation play an important role in the antifibrotic effects of pirfenidone.
TGF-β1 exerts biological activity by binding to the type I and II transmembrane receptors. In general, TGF-β1 and its receptor complex activate the Smad family to induce nuclear translocation and expression of a number of target genes. Although Smad proteins possess binding activities to their own recognizing elements in promoter regions, those affinities or productivities have been reported as insufficient for inducing transcription of related genes, ultimately requiring several cooperatively functioning transcriptional factors.Citation33,34 TGF-β1-stimulated expression of PAI-1 has been reported to be regulated by not only Smad signaling but also non-Smad signaling, including ERK.Citation35 Direct TGF-β1-inducible interaction between Smad and c-Jun has also been reported.Citation36 In addition, the present results further suggest that TGF-β1-induced Smad and MEK/ERK signaling pathways cooperatively regulate expression of profibrotic factors. Pirfenidone has been reported to attenuate TGF-β1-induced activation of MAPK in human lung fibroblasts.Citation37 In this study, pirfenidone inhibited TGF-β1-induced activation of ERK and c-Fos in rat proximal tubular epithelial cells. In this signal kinase experimental time period, transformation from epithelial cells to myofibroblasts induced by TGF-β1 was not reflected, findings that can be attributed only to epithelial cells. However, given that pirfenidone exerted no effects on expressions of α-SMA and vimentin, experimental results treated with pirfenidone would not be affected by EMT. Time-course observations showed that MEK inhibitor U0126 reduced ERK activity to below basal levels over the entire measurement period. In contrast, pirfenidone inhibited TGF-β1-induced activation of ERK without influencing the basal activity. These results suggest that pirfenidone acts on several cascades (e.g., TGF-β1 receptor or feedback mechanism)Citation38 activated by TGF-β1 without exerting a direct inhibitory effect on the MEK/ERK signaling kinase pathway, with further studies required to clarify the compound’s mechanism of action in detail.
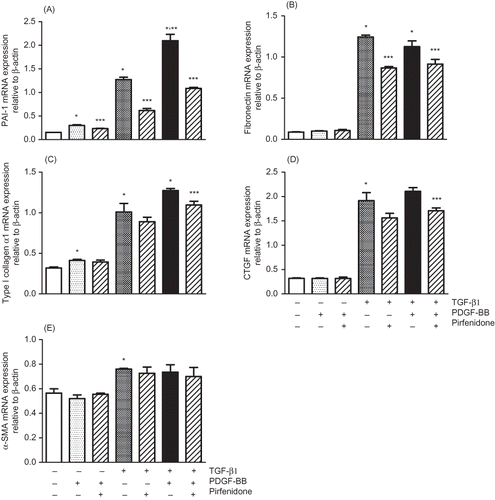
A number of factors have been reported to be involved in the development of renal fibrosis. Recent attention has been focused on the finding that expression of not only TGF-β1 but also PDGF, another key mediator of fibrosis in different organs including kidney, is increased coinciding with renal damage.Citation39,40 In TGF-β1-induced pulmonary fibrosis models, experimental introduction of PDGF as a recombinant protein or via viral vector results in fibrosis, which is in turn ameliorated by inhibitors of PDGF receptor tyrosine kinase activity.Citation22 In addition, the PDGF-B isoform has been shown to exert profibrotic effects in the tubulointerstitial compartmentCitation24 and mediate glomerular ECM accumulation in glomerulonephritis.Citation23 Pirfenidone treatment downregulated expression of IL-1β, a cytokine that induces fibroblasts to produce fibrogenic mediators such as PDGF and TGF-β1.Citation22 In this study, we demonstrated that PDGF-BB enhanced the expression of PAI-1, type I collagen, and CTGF with TGF-β1, while pirfenidone inhibited this cooperative effect. These present and previous findings suggest that TGF-β1 and PDGF may interact and work cooperatively to induce renal fibrosis, and that pirfenidone exerts its antifibrotic effect by inhibiting this interaction (). These factors (PAI-1, PDGF, and ECM) measured in this study are part of the presumed efficacy mechanism of pirfenidone, with other mechanisms/parameters also believed to play important roles in the compound’s antifibrotic effect; this calls for further studies to be conducted.Citation22
Figure 5. Pathogenetic pathways of renal fibrosis induced by TGF-β1 cooperative with PDGF and action point of pirfenidone indicated by the present results in cell-based assays.
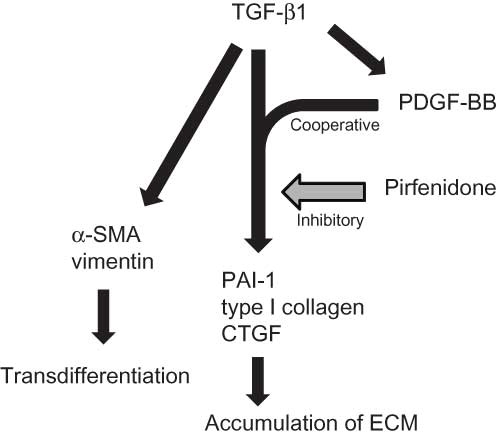
The molecular mechanism of renal fibrosis is believed to involve several factors, such as TGF-β1, PDGF, and PAI-1, all promoting the accumulation of ECM in the glomeruli and the interstitium of the cortex. These fibrotic factors also induce EMT, a process in which epithelial cells lose their phenotype and acquire myofibroblast-like characteristics that include greatly increased production of ECM components.Citation12 Thus, EMT plays an important role in renal fibrosis, its presence is becoming increasingly evident as chronic renal disease advances, and suppression of EMT is believed to offer protection against chronic kidney disease. Transdifferentiation from epithelial cells to myofibroblasts is generally defined based on representative changes in EMT markers, specifically by an increase in levels of mesenchymal markers (α-SMA and vimentin) but a decrease in those of epithelial markers (E-cadherin). The pathological role of TGF-β1 in EMT and sclerotic lesions has been well established and recognized as crucial.Citation10 In this study, TGF-β1 significantly increased α-SMA and vimentin expression (approximately double), strongly suggesting that TGF-β1 induces EMT in rat proximal tubular epithelial cells and prompting a shift to a higher ratio of myofibroblasts. Given that ECM is thought to be produced mainly by myofibroblasts, the increases in ECM expression induced by TGF-β1 and inhibition of expression by pirfenidone seen in this experiment are thought to be activities produced by myofibroblasts. In contrast, epithelial cells have also been reported to be capable of producing a variety of ECM proteins in response to TGF-β1,Citation41 suggesting that these effects contributed in both epithelial cells and transformed myofibroblasts. Previous reports have shown that, in the kidney interstitium, approximately 50% of the myofibroblasts are derived from local fibroblasts, 35% from epithelial cells via the EMT, and 15% from bone marrow-derived fibrocytes and mesenchymal cells.Citation42 In this study, we investigated the antifibrotic effects of pirfenidone in epithelial cells; only a portion of the antifibrotic effects induced by pirfenidone might have been examined. We found that pirfenidone exerted no significant effects on TGF-β1-induced increases in α-SMA and vimentin expression, suggesting that pirfenidone cannot inhibit TGF-β1-induced EMT. However, Yuan et al.Citation43 reported conflicting findings, noting that pirfenidone significantly suppressed TGF-β1-induced increases in mRNA expression of ECM and EMT markers in human proximal tubular epithelial cells. In addition, recent genetic lineage analysis has suggested that, contrary to widely accepted theories, epithelial cells may not transform to myofibroblasts during EMT.Citation44 Proper assessment of antifibrotic actions requires performing experiments and gathering data from not only in vitro assays but also in vivo experiments including animal models. As such, further detailed examination will be required to clarify whether pirfenidone suppresses TGF-β1-induced EMT in the kidney.
In summary, the present findings suggest that TGF-β1 closely correlates with renal fibrosis in cooperation with PAI-1 and PDGF in rat proximal tubular epithelial cells, with pirfenidone inhibiting these fibrosis cascades, thereby suggesting similar antifibrotic effects in the kidney. These findings may be clinically useful for preventing or ameliorating progressive renal diseases.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Notice of Correction
The Early Online version of this article published online on 24 September 2012 contained an error in Figure 4. The legend “Pirfenidone” and “PDGF-BB” in Figure 4D and E had been placed in the wrong order. The error has been corrected for this version.
Related Research Data
REFERENCES
- Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis. 2005;12:353–365.
- Boor P, Ostendorf T, Floege J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643–656.
- Negri AL. Prevention of progressive fibrosis in chronic renal diseases: Antifibrotic agents. J Nephrol. 2004;17:496–503.
- Declèves AE, Sharma K. New pharmacological treatments for improving renal outcomes in diabetes. Nat Rev Nephrol. 2010;6:371–380.
- Wight TN, Potter-Perigo S. The extracellular matrix: An active or passive player in fibrosis? Am J Physiol. 2011;301:G950–G955.
- Sharma SK, Ziyadeh FN. The emerging role of transforming growth factor-beta in kidney diseases. Am J Physiol. 1994;266:F829–F842.
- Gagliardini E, Benigni A. Therapeutic potential of TGF-beta inhibition in chronic renal failure. Expert Opin Biol Ther. 2007;7:293–304.
- Ma LJ, Fogo AB. PAI-1 and kidney fibrosis. Front Biosci. 2009;14:2028–2041.
- Eddy AA. Serine proteases, inhibitors and receptors in renal fibrosis. Thromb Hemost. 2009;101:656–664.
- Fan JM, Ng YY, Hill PA, . Transforming growth factor-beta regulates tubular epithlial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467.
- Burns WC, Kantharidis P, Thomas MC. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs. 2007;185:222–231.
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222.
- Fukasawa H, Yamamoto T, Suzuki H, . Treatment with anti-TGF-β antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-β signaling. Kidney Int. 2004;65:63–74.
- Taniguchi H, Ebina M, Kondoh Y, . Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829.
- Maher TM. Pirfenidone in idiopathic pulmonary fibrosis. Drugs Today. 2010;46:473–482.
- Macias-Barragan J, Sandoval-Rodriguez A, Navarro-Partida J, Armendariz-Borunda J. The multifaceted role of pirfenidone and its novel targets. Fibrogenesis Tiss Rep. 2010;3:16.
- Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998;54:99–109.
- Shimizu T, Fukagawa M, Kuroda T, . Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl. 1997;63:S239–S243.
- Takakura K, Fujimori A, Chikanishi T, . Renoprotective properties of pirfenidone in subtotally nephrectomized rats. Eur J Pharmacol. 2010;629:118–124.
- Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995; 125:779–785.
- Nakayama S, Mukae H, Sakamoto N, . Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008;82:210–217.
- Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97.
- Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23.
- Ostendorf T, Kunter U, Grone HJ, . Specific antagonism of PDGF prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol. 2001;12:909–918.
- Ostendorf T, Eitner F, The FJ. PDGF family in renal fibrosis. Pediatr Nephrol. 2011;26(9):2728–2731 (online).
- Yamate J. Heterogeneity of macrophage populations and myofibroblasts appearing in rat renal interstitial fibrosis. J Toxicol Pharmacol. 2007;20:185–195.
- Shimizu F, Fukagawa M, Yamauchi S, . Pirfenidone prevents the progression of irreversible glomerular sclerotic lesions in rats. Nephrology. 1997;3:315–322.
- Hammes MS, Lieske JC, Pawar S, Spargo BH, Toback FG. Calcium oxalate monohydrate crystals stimulate gene expression in renal epithelial cells. Kidney Int. 1995;48:501–509.
- Zhang G, Kim H, Cai X, Lopez-Guisa JM, Carmeliet P, Eddy AA. Urokinase receptor modulates cellular and angiogenic responses in obstructive nephropathy. J Am Soc Nephrol. 2003; 14:1234–1253.
- Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301.
- Ma J, Weisberg A, Griffin JP, Vaughan DE, Fogo AB, Brown NJ. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int. 2006; 69:1064–1072.
- Shihab FS, Bennett WM, Yi H, Andoh TF. Pirfenidone treatment decreases transforming growth factor-β1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am J Transplant. 2002;2:111–119.
- Ikushima H, Komuro A, Isogaya K, . An Id-like molecule, HHM, is a synexpression group-restricted regulator of TGF-β signaling. EMBO J. 2008;27:2955–2965.
- Schiller M, Dennler S, Anderegg U, . Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem. 2010;285:409–421.
- Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Hemost. 2008;100:976–983.
- Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;27:909–913.
- Ozes ON, Stevens SK, Phillips RJ, Blatt LM. Pirfenidone attenuates transforming growth factor-beta-induced activation of P38-alpha and P38-gamma mitogen-activated protein kinases in human lung fibroblasts: Implications for treatment of fibrotic lung diseases. Chest. 2006;130:114S (Abstract).
- Cirit M, Wang CC, Haugh JM. Systematic quantification of negative feedback mechanisms in the extracellular signal-regulated kinase (ERK) signaling network. J Biol Chem. 2010;285:36736–36744.
- Frazier KS, Paredes A, Dube P, Styer E. Connective tissue growth factor expression in the rat remnant kidney model and association with tubular epithelial cells undergoing transdifferentiation. Vet Pathol. 2000;37:328–335.
- Sommer M, Eismann U, Deuther-Conrad W, . Time course of cytokine mRNA expression in kidneys of rats with unilateral ureteral obstruction. Nephron. 2000;84:49–57.
- Creely JJ, Dimari SJ, Howe AM, Haralson MA. Effects of transforming growth factor-beta on collagen synthesis by normal rat kidney epithelial cells. Am J Pathol. 1992;140:45–55.
- Kisseleva T, Brenner DA. Mechanisms of fibrogenesis. Exp Biol Med. 2008;233:109–122.
- Yuan Q, Wang L, Zhang F, . Fluorofenidone suppresses epithelial-mesenchymal transition and the expression of connective tissue growth factor via inhibiting TGF-β/Smads signaling in human proximal tubular epithelial cells. Pharmazie. 2011;66:961–967.
- Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol. 2012;27:183–193.