Abstract
Objective: To construct a plasmid containing a urate oxidase and creatinine hydrolase fusion gene and transform the plasmid into Escherichia coli to decompose uric acid and creatinine. Methods: According to the GenBank data for the urate oxidase gene, specific primers were designed to amplify and remove the stop codon for the urate oxidase gene. The gene was then ligated into the plasmid pMG36e to construct pMG36e-U. Then, using the GenBank database for the creatinine hydrolase gene, primers were designed to amplify the creatinine hydrolase gene. This gene was ligated into pMG36e-U to form pMG36e-U/C. Next, this construct was transformed into E. coli, which was confirmed by screening the recombinant E. coli and sodium dodecylsulfonate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The engineered bacteria were cultured with a specific concentration of creatinine and uric acid for 24 h. Then, the concentrations of creatinine and uric acid in the culture fluid were measured. Results: The recombinant gene fragment was approximately 1.68 kb, and it contained the urate oxidase and creatinine hydrolase genes. The transformed E. coli expressed creatinine hydrolase and uric acid oxidase. The creatinine decomposition rate increased by 43.5%, and the uric acid decomposition rate increased by 42.32%. Conclusion: The constructed recombinant plasmid containing a fusion gene of creatinine hydrolase and uric acid oxidase was transformed into E. coli, and the enzymatic activities were expressed.
INTRODUCTION
To date, chronic renal failure has been treated using hemodialysis, peritoneal dialysis, or kidney transplantation. The cost of these treatments is high, and these treatments have difficulty in treating the complications of chronic renal failure. Therefore, there is a need for a noninvasive, cost-effective dialysis therapy. Intestinal bacterial therapy, including “natural strains or genetically engineered bacteria,” has been used to metabolize and remove toxins from the blood to treat chronic renal failure and has drawn attention in recent years. Uric acid and creatinine are the highest concentrations of toxins in the blood of patients with chronic renal failure.
Some bacteria have high urate oxidase and/or creatinine hydrolase activity.Citation1,2 If these genes were cloned, the genetically engineered bacteria could be used as an oral probiotic to treat chronic renal failure by metabolizing and removing the uremic toxins. This treatment would have economic, social, and environmental benefits.
MATERIALS AND METHODS
Materials
Candida ulitis and Pseudomonas putida were provided by the China General Microbiology Culture Center. The pMG36e expression vector was a gift from Dr. J. Kok (University of Groningen in the Netherlands). The pMD-18T vector was purchased from the Takara Company (Japan). The Escherichia coli strain DH5α was made in our laboratory. The restriction enzymes SacI, XbaI, and Hind III were purchased from the Takara Company. The DNA Ligation Kit was purchased from the Takara Company. Sodium dodecylsulfonate (SDS) was purchased from Sigma (Sigma-Aldrich (Shanghai) Trading Co. Ltd., Shanghai, China). The primers were made by Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China. The Gene Ruler DNA ladder and the MBI λ-Hind III digest marker were purchased from the Takara Company (Takara Biotechnology (Dalian) Co. Ltd., Dalian, China).
Methods
Construction of the recombinant plasmid pMG36e-U
Based on previous reports, the following primers were designed to amplify the C. ulitis uricase gene (GenBank sequence No. E12709, nt1–nt912) and remove the stop codon: Primer-1, 5′AGAGCTCCCATGAGCAAGA GTGTTTTTGT3′; Primer-2, 5′AATCTAGACAAC TTGGTCTTCTCCTTAC3′. The uricase gene 5′ primer (Primer-1) had the SacI restriction site, and the 3′ primer (Primer-2) had an XbaI restriction site. For the polymerase chain reaction (PCR) amplification, the reaction conditions were 95°C for 30 s, 60°C for 30 s, and 72°C for 60 s for a total of 25 cycles. The PCR product was ligated into the pMD18-T vector to form the recombinant plasmid pMD18-TU, which was transformed into E. coli DH5α. The positive clones were picked, sequenced, and digested using SacI and XbaI. The resulting gene fragment was purified and ligated into the plasmid pMG36e using the same restriction enzymes. The resulting construct was transformed into E. coli DH5α, and the clones were screened for expression.
Building the recombinant plasmid pMG36e-U/C
Based on previous reports, the following primers were designed to amplify the P. putida creatinine hydrolase gene (GenBank sequence D45424, nt41-nt823): Primer-3, 5′AATCTAGACAACTTGGTCTTCTCC TTAC3′; Primer-4, 5′AGAAGCTTTCAGGTGGGC GGGAACTCCT3′. The creatinine hydrolase gene 5′ primer (Primer-1) had an XbaI restriction site, and the 3′ primer (Primer-2) had a Hind III restriction site. The PCR product was ligated into the pMD18-T vector to form pMD18-TC, which was transformed into E. coli DH5α. The positive clones were picked, sequenced, and digested using XbaI and Hind III. The resulting gene fragment was purified and ligated into the plasmid pMG36e-U using the same restriction enzymes. The resulting construct pMG36e-U/C was transformed into E. coli DH5α, and the clones were screened for expression using sodium dodecylsulfonate-polyacrylamide gel electrophoresis (SDS-PAGE).
Genetically engineered bacteria decompose uric acid and creatinine in vitro
The E. coli expressing pMG36e-U/C were inoculated 1:25 in De Man, Rogosa and Sharpe medium (MRS) medium, cultured at 37°C, then washed in sterile saline and centrifuged three times, and resuspended in normal saline. Using the Maxwell turbidimetric method, the cell count was adjusted to approximately 1.5 × 108 cfu/mL. For every 200 μL of bacteria, 800 μL of solution containing specific concentrations of creatinine and uric acid was added. The bacteria were cultured at 37°C for 24 h and the culture was centrifuged at 40,000 rpm/min. Then, the cells were centrifuged for 5 min, and the concentrations of creatinine and uric acid were measured using a biochemical analyzer. For the control, 200 μL of normal saline was added to five tubes containing the above medium . To calculate the decomposition rates of creatinine and uric acid, the following formula was used: decomposition rate (%) = (the creatinine or uric acid content of the control group – the content of the intervention group)/the creatinine or uric acid content of the control group.
RESULTS
Using Primer-1 and Primer-2 for PCR, the C. ulitis urate oxidase gene was amplified, purified, and ligated into the pMD18-T vector to construct pMD18-TU. This plasmid was transformed into E. coli, which were screened. The recombinant plasmid was extracted, sequenced, and double digested using SacI and XbaI. The sequencing results were consistent with the GenBank sequence for the uric acid oxidase, as shown in . The M lane has a molecular weight marker. Lane 1 is a control double digestion of the empty vector pMD18-T using SacI and XbaI. Lanes 2–4 represent double digestions of p-MD18-TU using SacI and XbaI. The gene fragment size was approximately 0.9 kb.
Figure 1. Plasmid extraction and digestion of the recombinant E. coli expressing pMD18-TU. M—GeneRuler DNA ladder marker. Lane 1—pMD18-T digested using SacI and XbaI to serve as a control. Lanes 2–4—pMD18-TU digested using SacI and XbaI, which yielded a 0.9 kb gene fragment.
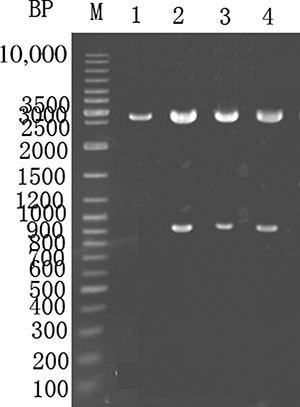
PCR was used to remove the uricase gene stop codon, followed by SacI and XbaI double digestion and ligation into plasmid PMG36e to form pMG36e-U, which was transformed into E. coli DH5α. The positive clones were screened using Luria-Bertani medium (LB) containing neomycin, as shown in . Lanes 1–3 show a PCR product of approximately 0.9 kb in length. Lane 4 shows a PCR using the original E. coli as a control, and no PCR was observed.
Figure 2. PCR recombinant E. coli (PCR identification of recombinant E. coli) which is 0.9 kb in size. M—GeneRuler DNA ladder marker. Lanes 1–3—the recombinant E. coli were used for PCR amplification of the gene fragment using the pMG36e-U template. Lane 4 serves as a control, and the original E. coli were used for PCR.
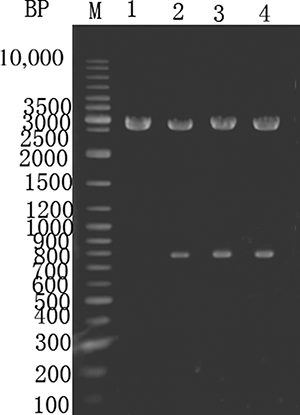
The P. putida creatinine hydrolase gene was PCR amplified, purified, and ligated into the pMD18-T vector to construct pMD18-TC and transformed into E. coli. The clones were screened, double digested using XbaI and Hind III, and sequenced. The sequence matched the GenBank database for the creatinine hydrolase gene, as shown in . Lane 1 shows a control digestion of pMD18-T using XbaI and Hind III. Lanes 2–4 show an approximately 0.78 kb fragment after XbaI and Hind III double digestion of pMD18-TC.
Figure 3. Enzyme digestion of E. coli expressing pMD18-TC. M—GeneRuler DNA ladder marker. Lane 1—pMD18-T digested with XbaI and Hind III. Lanes 2–4—pMD18–T-C digested with XbaI and Hind III.
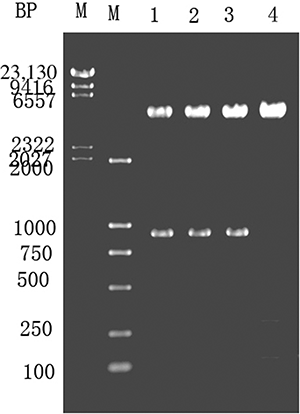
The creatinine hydrolase gene was PCR amplified, ligated into pMG36e-U/C using XbaI and Hind III to form pMG36e-U/C, and transformed into E. coli DH5α. The clones were screened using LB containing erythromycin, as shown in . Lanes 1–3 reveal a 1.68 kb fragment after SacI and Hind III digestion of the recombinant plasmid. Lane 4 shows a control digestion of pMG36e using SacI and Hind III.
Figure 4. Enzyme digestion of E. coli expressing pMG36e-U/C. M—λ-Hind III digest marker. Lanes 1–3—pMG36e-U/C digested with SacI and Hind III. Lane 4—pMG36e digested with SacI and Hind III.
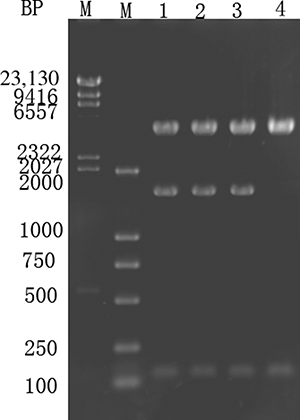
Figure 5. Purified recombinant urate oxidase according to SDS-PAGE (12%) analysis. Lane M—a low molecular weight protein ladder. Lane 1—the crude lysate from the recombinant Lactobacillus expressing urate oxidase (Lb-PMG36e-U). Lane 2—recombinant uricase purified using diethylaminoethyl (DEAE) DE52.
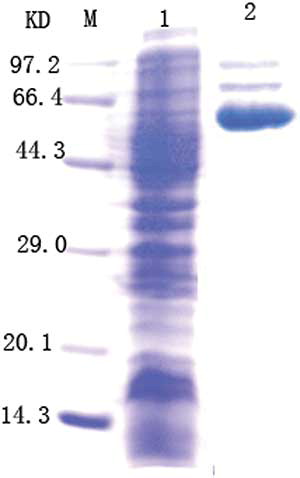
Recombinant E. coli containing pMG36e-U/C were cultured in medium containing uric acid and creatinine and sonicated for SDS-PAGE analysis of the crude enzyme lysate. The results are shown in .
The decomposition rates for uric acid and creatinine in genetically engineered bacteria were measured as follows.
The mean creatinine concentration was 1008.9 ± 18 umol/L in the control group and 768 ± 19.7 umol/L in the experimental group, indicating that the creatinine decomposition rate increased by 43.5% in the experimental group. The mean uric acid concentration was 180.9 ± 18 mmol/L in the control group and was 120.1 ± 17 mmol/L in the experimental group, indicating that the uric acid decomposition rate increased by 42.32% in the experimental group.
DISCUSSION
In recent years, following digestive enzyme therapy, intestinal bacterial therapy has become a new method to remove uremic toxins. In digestive enzyme therapy, microencapsulated, uremic toxin decomposition enzymes, urease and uricase, have been delivered to the digestive tract to break down uremic toxins. However, an enzyme can only break down a specific substrate, and a variety of uremic toxins accumulate in patients with chronic renal failure. Therefore, the use of natural bacteria, which have a low toxicity and can produce a variety of enzymes to break down urinary toxins to treat renal failure has gained attention.
Ranganathan et al.Citation3 have isolated a species of bacteria from the intestine that can break down a variety of uremic toxins. An in vivo study found that this toxin clearance ability was similar to hemodialysis. However, this bacteria, which carry resistance to antibiotics, lead to overgrowth and severe infection in the intestines, which may be problem for patient safety. Therefore, people have investigated using genetically engineered bacteria to clear urinary toxins. The cloning of high activity of the uremic toxin decomposition enzymes into bacteria to be used as probiotics has gained attention.
An oral preparation of the probiotics could be made to enter the digestive tract and decompose uremic toxins. So far, engineered bacteria have only been able to express a single enzyme, such as ureaseCitation4 or uricase.5 However, the main biochemical feature of chronic renal failure is the accumulation of a variety of urinary toxins. Removing a variety of urinary toxins using bacteria engineered with a single enzyme has been difficult. Therefore, studies have tried to clone the genes of the several uremic toxin enzymes into bacteria. According to the literature, C. ulitis yeast has a high-activity uric acid oxygenase,6 and P. putida has a high-activity creatinine hydrolase. Our previous work has been to design genetically engineered bacteria to break down uric acid and creatinine.7,8 Thus, we cloned the uric acid oxidase gene and creatinine hydrolase genes into a plasmid. We selected the plasmid pMG36e, which is a common expression vector that can be replicated in E. coli, lactic acid bacteria, and Bacillus subtilis. The plasmid size is 3.6 kb, which includes an erythromycin resistance gene, P32 promoter, polyclonal point, and a “prtp” transcription terminator, and has good genetic stability. This plasmid has been used to express a variety of exogenous genes. Thus, pMG36e plays an important role in recombinant DNA technology.
Candida ulitis is a eukaryote, and its urate oxidase gene does not have an intron. We designed specific primers based on the known sequences for the uric acid oxidase and creatinine hydrolase genes to amplify the genes and remove the stop codon from the urate oxidase gene. This uric acid gene was ligated into the sequencing plasmid pMG36e to form pMG36e-U. Then, the creatinine hydrolase gene from P. putida was ligated into pMG36e-U to make pMG36e-U/C, which was transformed into E. coli. Escherichia coli was selected as the bacteria strain for the following reasons. (1) Escherichia coli is the most important and abundant bacteria in the large intestine and is a normal inhabitant in the intestines of humans and animals. During breastfeeding at birth, E. coli colonizes the intestines. Escherichia coli can inhibit the growth of the microorganisms that break down proteins and reduce the amount of protein decomposition products in human intestines. Additionally, E. coli can synthesize vitamins B and K. (2) Escherichia coli is an easy-to-use heterologous gene expression host with a clear genetic background and is simple to culture and use. Escherichia coli is the most successful gene expression host and the most widely used. Many genetic engineering experts have used E. coli. (3) Some studies have shown that microencapsulated, genetically modified E. coli DH5 could reduce the concentrations of urea, uric acid, creatinine, and some inorganic molecules in in vitro and in vivo (rat) chronic renal failure experiments. Escherichia coli may be a candidate for bacterial therapy for chronic renal failure (CRF).9 (4) Escherichia coli is an opportunistic pathogen and is not pathogenic in the intestine. Our results suggest that E. coli not only expressed the recombinant plasmid but also promoted uremic toxin decomposition. There are broad applications for the biological decomposition of urine toxins.
Declaration of interest: The authors report no conflict of interest. The authors are solely responsible for the content and writing of this article.
This study was supported by a specialized Research Fund from the Natural Science Foundation (Grant No. 30871168).
REFERENCES
- Zhang J, Ren J, Li B, . Construction, expression, purification and characterization of mutant of Aspergillus flavus urate oxidase. Sheng Wu Gong Cheng Xue Bao. 2010;26(8):1102–1107.
- Yamashita K, Nakajima Y, Matsushita H, . Substitution of Glu122 by glutamine revealed the function of the second water molecule as a proton donor in the binuclear metal enzyme creatininase. J Mol Biol. 2010;396(4):1081–1096.
- Ranganathan N, Patel BG, Ranganathan P, . In vitro and in vivo assessment of intraintestinal bacteriotherapy in chronic kidney disease. Am Soc Art Int org. 2006;52(1):70–79.
- Zhang S, Li D, Tian K, . Development of a recombinant ureolytic Lactococcus lactis of urea removal. Artif Cells Blood Substit Immobil Biotechnol. 2009;37(6):227–234.
- Liu G-Z, Chen J-H, Wang M. Screening of uricase producing strains, cloning and expression of uricase gene. J China Pharm Univ. 2005;36(5):464–466.
- Safra N, Ling GV, Schaible R, .Exclusion of urate oxidase as a candidate gene for hyperuricosuria in the Dalmatian dog using an interbreed backcross. J Hered. 2005;96(7):750–754.
- Cheng X, Zhang Y-X, Yang B, .Construction of gene engineering bacterium to produce urate oxidase. Central South Pharmacy 2009;7(6):401–404.
- Cheng X, Zhang Y-X, Zhou G-F. Production of creatinine hydrolase genetically engineered bacteria. J Biomed Eng Res. 2009;28(4):299–302.
- Jain P, Shah S, Coussa R, . Potentials and limitations of microorganisms as renal failure biotherapeutics. Biologics. 2009;3:233–243.