
Abstract
Acute kidney injury (AKI) is one of the most common complications in patients with severe sepsis. The development of septic AKI increases patients’ mobility and even mortality. Toll-like receptor 2 (TLR2), as a membrane surface receptor for bacterial, fungal, viral and certain endogenous substances, has been described to contribute to the development of septic AKI; however, the renal cell types associating TLR2 overactivation in septic AKI has not been described. In the current study, we investigated the TLR2 activation patterns in the kidney of lipopolysaccharide-induced septic AKI mice. Our results demonstrated that mRNA level of TLR2 significantly increased in the kidney of lipopolysaccharide-treated mice. Immunohistochemistry revealed the overactivation of TLR2 in the glomeruli. Double immunofluorescence analysis shows the precise distribution of TLR2 by showing the colocalization of TLR2 in glomeruli with synaptopodin, a podocyte marker, and Tie2, an endothelial marker. In addition, proapoptotic molecules Bax and Caspase-3 were increased in the glomeruli of lipopolysaccharide-treated mice. Together, the current study indicates that TLR2 is overactivated in the glomerular endothelial cells and podocytes in septic AKI mice, while the abundance of Bax and Caspase-3 were increased in the glomeruli of these mice, it may supply a clue that TLR2 induced these cell apoptosis in AKI. This finding provides an alternative mechanism to understand AKI development and potential targets for treatment.
Introduction
Acute kidney injury (AKI), a common entity in critically ill patients, is mostly triggered by severe sepsis.Citation1–3 Of patients with septic AKI, about 30% need renal replacement therapy.Citation4 Given the severity of this disease, much work has been done in an effort to understand the mechanisms underlying septic AKI in addition to the strategy improvement in septic AKI treatment. However, after decades of research, little is known about the pathology of septic AKI. Up to now, it is believed that the alteration in kidney hemodynamics and/or other non-hemodynamic factors, such as immunological factors might be two possible contributing factors to the establishment of this disease.Citation2,Citation3,Citation5
Through either of these two mechanisms, the recognition of bacterial products by toll-like receptors (TLRs) is involved in the kidney damage after sepsis. TLRs, a family of transmembrane proteins, are the major pattern recognition receptors (PRRs) binding to molecules that are broadly shared by pathogens but distinguishable from host molecules, namely pathogen-associated molecular patterns (PAMPs). TLRs play a central role in innate immune system.Citation6 The activation of TLRs is responsible for the inflammatory responses in sepsis. Moreover, evidence have showed that over activation of TLRs, especially TLR2, contributes to the development of septic AKI.Citation7,Citation8 However, the cells with TLR2 overactivation in septic AKI are yet to be identified. TLR2 can trigger the expression of proinflammatory cytokines upon the activation by binding to lipopeptides from bacteria.Citation9–11 In the current study, we investigated the activation pattern of TLR2 in septic AKI using lipopolysaccharide (LPS)-induced septic AKI mice as a model. The results of the study could identify cell subsets with TLR2 overactivation in septic AKI, consequently providing targets for improved septic AKI treatment development.
Materials and methods
Animals
Six- to eight-week-old male BALB/c mice weighing 20–22 g were bought from SLAC Laboratory Animal Center and housed in a specific pathogen-free (SPF) environment with food and water supplied. This study was approved by the Bioethics Committee of the First People’s Hospital of Kunshan and performed in accordance with the guidelines of Laboratory Animal Science Association.
Murine septic AKI model
For the induction of murine septic AKI model, mice were intraperitoneally injected LPS (10 mg/kg, L2880, Sigma-Aldrich, St. Louis, MO) as previously described.Citation12 Twenty-four hours after LPS injection, mice were sacrificed.
Sampling
Urine samples were collected for a 24 h period following LPS injection. Twenty-four hours after LPS treatment, mice were sacrificed and blood and kidney samples were collected. Samples from age-matched normal mice were also collected and used as control.
Assessment of renal function
Sera were obtained from blood samples by centrifugation (1600g, 25 min at 4 °C). Blood urea nitrogen (BUN) and the serum creatinine (SCr) levels were determined by Hitachi 7060 automated Chemistry Analyzer (Diamond Diagnostics, Holliston, MA) according to the manufacturer’s instructions. Proteinuria (mg/mL) and uric creatinine concentration (mg/mL) were measured with the appropriate kits from Advia Chemistry 1650 (Bayer Healthcare AG, Leverkusen, Germany).
RNA extraction and real-time PCR
Total RNA was extracted from kidneys using TRIzol reagent (Invitrogen, Waltham, MA) according to the manufacturer’s instructions. Total RNA (3–5 μg) was then transcribed into cDNA by Superscript II reverse transcriptase (Invitrogen) and Oligo(dT)18 primers (Invitrogen). Real-time quantitative PCR was performed with an iCycler iQ™ Real-Time PCR Detection System and the Absolute QPCR SYBR Green premix (Takara, Shiga, Japan). Primers used for TLR2 amplification were:Citation13 forward, 5′ CAA ATG GAT CAT TGA CAA CAT CATC 3 reverse, 5′ TTC GTA CTT GCA CCA CTC GC 3 Primers used for β-actin were:Citation14 forward, 5 TGT TAC CAA CTG GGA CGA CAT GGA 3′; reverse, 5′ CCG CTC GTT GCC AAT AGT GAT GAC 3′. Expression levels of TLR2 were normalized to those of β-actin in the same samples using the 2−ΔΔCt method.
Histology
Kidney sections from LPS and control mice were incubated for 16 h in 4% paraformaldehyde, and subsequently dehydrated, paraffin embedded and stained with periodic acid-Schiff reagent (PAS). Ten kidney sections per mouse (n = 10 mice for each group) were analyzed and 20–30 glomeruli per kidney sections were randomly checked under microscope.
Immunohistochemistry analysis
For immunohistochemistry (IHC) study, kidney sections were first dewaxed and rehydrated and then antigen retrieval was performed by immersing the slides in boiling 0.01 mol/L citrate buffer in a 500 W microwave oven for 15 min. The endogenous peroxidase and non-specific spots were blocked with 0.3% H2O2 (30 min) and normal blocking serum (20 min), respectively. Following blocking, slides were washed with PBS and incubated overnight at 4 °C with rabbit anti-mouse TLR2 (Sigma-Aldrich), Bax (Santa Cruz Biotechnology, Santa Cruz, CA) and Caspase-3 (Abcam, Cambridge, MA), respectively. After extensive washes with PBS, slides were then incubated with biotinylated goat anti-rabbit antibodies (R&D Systems) and avidin-biotinylated horseradish peroxidase complex (Vector Laboratories) for 1 h and 15 min at room temperature, respectively. The colorimetric reaction was developed by diaminobenzidine (Sigma-Aldrich) for 10 min.
Double immunofluorescence labeling analysis
Double immunofluorescence labeling studies on kidney tissues were performed with 4 µm-thick cryostat sections, fixed in acetone for 10 min, air-dried 30 min at room temperature, and then kept in PBS for 5 min and blocked in 5% BSA-PBS. The slides were simultaneously incubated with rabbit anti-TLR2 antibody and mouse anti-synaptopodin antibody (R&D Systems) or rat anti-tie2 antibody (Progen Biotecknik GmbH). After washing with PBS, the slides were simultaneously incubated with fluorescein isothiocyanate-cogitated goat anti-rat IgG or goat anti-mouse IgG and cyanine 3-conjugated goat anti-rabbit IgG. Sections were examined by fluorescence microscopy (Carl zeiss, Oberkochen, Germany) using red and green filters.
Statistical analysis
The data are expressed as mean ± standard deviation (SD). Student’s t-test was applied for statistical comparisons and a p value less than 0.05 was considered statistically significant. All analyses were performed with SPSS 10.0 (SPSS, Chicago, IL).
Results
Induction of septic AKI mice with LPS treatment
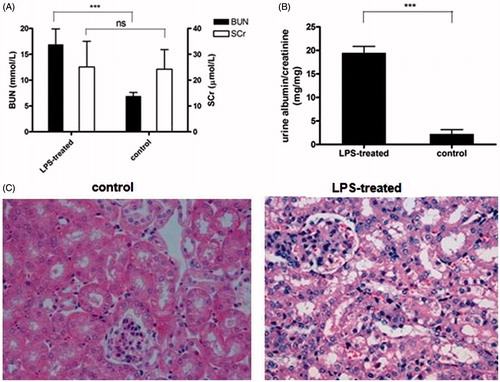
The septic AKI mice were obtained by LPS treatment for 24 h. After treatment, the renal function of treated mice was determined. As shown in , BUN and urinary albuminuria levels were considerably higher in LPS-treated mice than in healthy control mice, indicating that declined renal function was induced in LPS-treated mice. Moreover, histologic analysis by light microscopy also revealed that there was morphological evidence of glomeruli abnormality, proximal tubular epithelial cell injury, interstitial edema and interstitial inflammation in renal sections from LPS-treated mice ().
Figure 1. Induction of septic AKI mice with LPS treatment. (A) Serum urea and creatinine (SCr) were measured 24 h after LPS injection. Data shown as means ± SD of three independent experiments (p = 0.00071). Urine albumin to creatinine ratio was significantly increased in mice treated with LPS compared with healthy control mice. (19.51 ± 1.35 compared to 2.25 ± 0.9, p = 0.00067) (Upr/Ucr mg/mg). (C) Kidney sections was stained with PAS staining and checked under light microscope. Data shown here are one out of three independent experiments.
The overactivation of TLR2 was located in the glomerular endothelial cells and podocytes
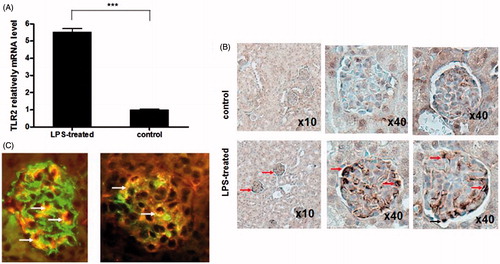
TLR2 over activation has been reported to be involved in septic AKI development,Citation7,Citation8 however, the expression pattern of TLR2 has not been described. Consequently, we next determined the cells with TLR2 overactivation in the kidney of septic AKI mice. First, we measured the TLR2 mRNA level in the whole kidney of septic AKI mice. Our results showed that TLR2 expression in kidney was significantly increased 24 h after LPS treatment, which was consistent to the previous descriptions ().Citation7,Citation8 To further identify the over expressing TLR2 in the kidney after septic AKI, we next determined the expression of TLR2 by immunohistochemistry (IHC), and revealed the overexpression of TLR2 in glomerular resident cells. Double immunofluorescence labeling indicated the colocalization of TLR2 in the glomeruli with synaptopodin, a podocyte marker and Tie2, an endothelial marker. As shown in , our results revealed that the TLR2 expression after LPS treatment was mainly located in the glomerular endothelial cells and podocytes.
Figure 2. The overactivation of TLR2 in septic AKI mice kidney. (A) TLR2 mRNA level was assessed by realtime PCR (n = 10). Data shown as means ± SD of three independent experiments (p = 0.00082). (B) Distribution of TLR2 overactivation in the kidney of LPS-treated mice. TLR2 expression was detected by IHC analysis and visualized under light microscope. The overactivation of TLR2 was located in the glomerular resident cells: endothelial cells (arrow heads) and podocytes (arrow heads) in septic AKI mice.(C) Double immunofluorescence analysis to precise the distribution of TLR2 in the glomeruli, double labelings were shown that TLR2 (arrow heads) partially colocalized with synaptopodin a podocyte marker (left) and Tie2, an endothelial marker (right), as arrows shown. Data shown here are at least one out of three independent experiments.
Increased Bax and Caspase-3 expression in the kidney of septic AKI mice
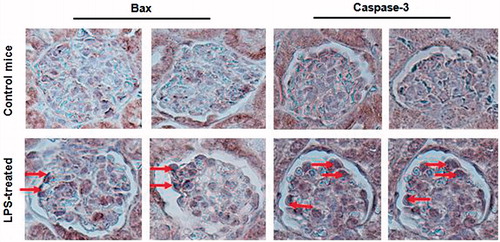
It has been described by recent study that apoptosis was involved in septic AKI.Citation15 To investigate whether the activation of TLR2 is related to the cell apoptosis, we studied the distribution of Bax and Caspase-3 in the kidney of septic AKI mice. Our results demonstrated that high levels of Bax and Caspase-3 were detected in the LPS-treated mice glomerular cells along the external side of some capillary loops (). These results implied that overexpression of TLR2 in glomeruli might involve in glomerular cells apoptosis. Further, inhibition of TLR2 by pharmacological inhibitors of TLR2 signaling or silencing of TLR2 by SiRNA need to be performed to determine whether inhibited TLR2 activation may prevent the induction of apoptosis in septic AKI mice.
Figure 3. Increased Bax and Caspase-3 expression in the kidney of septic AKI mice. Bax (left) and Caspase-3 (right) expression was assessed by the IHC analysis and visualized under light microscope. Increased Bax and Caspase-3 expression in the kidney of septic mice (arrow heads).Data shown here are at least one out of three independent experiments.
Discussions
Septic AKI is a common problem in critically ill patients, which would increase the morbidity as well as mortality of patients. With decades of research, the mechanism underlying septic AKI has not been fully understood. As the main cause for AKI in hospitalized patients, sepsis is predominately due to Gram-negative bacteria.Citation16 LPS, a component of the outer membrane of Gram-negative bacteria, has been reported to be involved in sepsis development through activation of TLR inflammatory signaling pathways.Citation17,Citation18 It has been described previously that TLR2 overactivation contributed to the development of septic AKI,Citation7,Citation8 however, the exact mechanism remains unknown. In the current study, we explored the TLR2 expression patterns in the LPS-induced septic AKI mice model. Our results, for the first time, revealed that TLR2 was overactivated in the glomerular endothelial cells and podocytes of the kidney in septic AKI mice. Moreover, we further found out that the proapoptotic molecules Bax and Caspase-3 were increased in the glomeruli of LPS-treated mice, imply a potential relationship between TLR2 induction and cellular apoptosis in septic AKI mice. Although further investigation is needed, our study provided not only valuable information for the full understanding of TLR2-related septic AKI, but also potential targets for the treatment development against septic AKI.
To induce septic AKI mice, in the current study, we adopted the LPS injection strategy. LPS, one of the exogenous ligands for TLR2, has been widely used in sepsis research. LPS administration induces systemic inflammation that mimics many of the initial clinical features of sepsis and causes renal injury, including decreased GFR, increased blood urea nitrogen (BUN), increased renal neutrophil infiltration and albuminuria.Citation12,Citation19–21 In our study, declined renal function was successfully induced in the LPS-treated mice as indicated by increased BUN and urinary albuminuria to creatinine ratio levels. Although solid data have obtained from the classic septic AKI mice model that TLR2 is overactivated in the glomerular endothelial cells and podocytes of the kidney, it is still required to determine whether the results are universal or LPS-induced septic AKI-specific.
It is known that innate pathogen recognition and danger signaling are not restricted to immune cells. As a necessary signaling component in the innate immune system, TLR2 has also been reported to express on many cell types besides immune cells. Wolfs and colleagues showed that TLR2 are constitutively expressed on the renal epithelial cells of distal and proximal tubules, the epithelium of Bowman’s capsule, glomerular and endothelial cells in the I/R model kidney.Citation18 Pawar et al.Citation22 found that bacterial lipopeptide activated TLR2 expressed on cultured podocytes and glomerular endothelial cells in vitro. In our current study, we demonstrated that TLR2 was also overactivated in two non-immune cell types, podocytes and glomerular endothelium cells, in vivo using a LPS-induced AKI model.
Recently, the role of apoptosis in the pathogenesis of sepsis has also been studied. Administration of the Caspase inhibitors or overexpression of the anti-apoptotic protein Bcl-2 significantly improves the survival of septic mice.Citation23–25 Our study showed that apoptotic molecules Bax and Caspase-3 were highly expressed in the LPS induced AKI mice glomerular cells. Albeit detailed mechanisms are remained to be further defined, our study herein indicates that TLR2 overactivation in the podocytes and glomerular endothelium cells might involve in the apoptosis of glomerular cells. Inhibition of TLR2 by pharmacological inhibitors of TLR2 signaling or silencing of TLR2 by SiRNA need to be performed to determine whether inhibition of TLR2 activation may prevent the induction of apoptosis in septic AKI mice.
This study has some limitations, such as the duration of the experimental time course. Although the experimental time is just 24 h, it is enough to reproduce the initial phases of septic acute kidney injury. Another limitation of our study is that the results just depended on the IHC results and RT-PCR without more direct results, such as western blotting and experiments in vitro. Experiments are ongoing in our laboratory to clarify these points. Furthermore, the specific pathway of TLR2 activating in these two cells of kidney deserves further study.
In summary, our data indicate that TLR2 is overactivated in the glomerular endothelial cells and podocytes in septic AKI mice and it might be related to cell apoptosis. The results of the study provide not only valuable information for the complete understanding of TLR2-related septic AKI, but also potential targets for the treatment development against septic AKI.
Acknowledgments
We would like to thank Dr Shaoyu Zhang for helpful discussions and technical assistance in our studies.
Declaration of interest
The authors do not have any conflicts of interests.
References
- Bellomo R, Wan L, Langenberg C, et al. Septic acute kidney injury: New concepts. Nephron Exp Nephrol. 2008;109:e95–e100
- Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251
- Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442
- Thakar CV, Christianson A, Freyberg R, et al. Incidence and outcomes of acute kidney injury in intensive care units: A veterans administration study. Crit Care Med. 2009;37:2552–2558
- Wan L, Bagshaw SM, Langenberg C, et al. Pathophysiology of septic acute kidney injury: What do we really know? Crit Care Med. 2008;36:S198–S203
- Arslan F, Keogh B, McGuirk P, et al. TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm. 2010;2010:704202
- Gluba A, Banach M, Hannam S, et al. The role of Toll-like receptors in renal diseases. Nat Rev Nephrol. 2010;6:224–235
- Alves-Filho JC, Freitas A, Souto FO, et al. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci USA. 2009;106:4018–4023
- Liang MD, Bagchi A, Warren HS, et al. Bacterial peptidoglycan-associated lipoprotein: A naturally occurring toll-like receptor 2 agonist that is shed into serum and has synergy with lipopolysaccharide. J Infect Dis. 2005;191:939–948
- Zahringer U, Lindner B, Inamura S, et al. TLR2 – promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology. 2008;213:205–224
- Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. 2012;3:79
- Knotek M, Rogachev B, Wang W, et al. Endotoxemic renal failure in mice: Role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int. 2001;59:2243–2249
- Castoldi A, Braga TT, Correa-Costa M, et al. TLR2, TLR4 and the MYD88 signaling pathway are crucial for neutrophil migration in acute kidney injury induced by sepsis. PLoS One. 2012;7:e37584
- Lorenz E, Chemotti DC, Vandal K, et al. Toll-like receptor 2 represses nonpilus adhesin-induced signaling in acute infections with the Pseudomonas aeruginosa pilA mutant. Infect Immun. 2004;72:4561–4569
- Messaris E, Memos N, Chatzigianni E, et al. Apoptotic death of renal tubular cells in experimental sepsis. Surg Infect (Larchmt). 2008;9:377–388
- Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169
- Armstrong L, Medford AR, Hunter KJ, et al. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136:312–319
- Wolfs TG, Buurman WA, van Schadewijk A, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168:1286–1293
- Cunningham PN, Wang Y, Guo R, et al. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–2635
- Tiwari MM, Brock RW, Megyesi JK, et al. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: Role of nitric oxide and caspases. Am J Physiol Renal Physiol. 2005;289:F1324–F1332
- Xu C, Chang A, Hack BK, et al. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014;85:72–81
- Pawar RD, Castrezana-Lopez L, Allam R, et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll-like receptor 2 at the glomerular filtration barrier. Immunology. 2009;128:e206–e221
- Hotchkiss RS, Chang KC, Swanson PE, et al. Caspase inhibitors improve survival in sepsis: A critical role of the lymphocyte. Nat Immunol. 2000;1:496–501
- Oberholzer C, Oberholzer A, Clare-Salzler M, et al. Apoptosis in sepsis: A new target for therapeutic exploration. FASEB J. 2001;15:879–892
- Hotchkiss RS, Tinsley KW, Swanson PE, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96:14541–14546