
Abstract
While many previous studies have reported an association between the single-nucleotide polymorphisms (SNPs) of the podocin and proteinuria occurred, a conclusive relationship has not been defined in every oligoallelic state of amino acid (AA) mutations in podocin. In this study, we performed a meta-analysis of the published data to investigate the impact of the oligoallelic AA mutations of the podocin on proteinuria; a total 16 AA mutations were investigated for oligoallelic pathogenicity. Despite significant heterogeneity within some of the comparisons, the results revealed significantly higher risks of proteinuria in early-onset (onset age <16) individuals for five mutations (P118L, R138Q, R168H, V180M, and V260E), and in all onset ages individuals for five mutations (R138Q, G140X, R229Q, V260E, and V290M) compared to non-variant individuals. We also tested the steroid response in individuals with R229Q and E237Q. No statistically significant differences in the two mutations carrier rate were observed between steroid resistance patients and controls. No AA mutation was selected for meta-analysis on the recurrence of proteinuria after renal transplantation as lack of control data. In conclusion, our meta-analysis tested the pathogenicity of the oligoallelic AA mutations in podocin and suggested the potential causative mutations, and the alleles showing an association with protein susceptibility. The sensitivity and specificity of each causative mutation are pending further testing.
Introduction
Proteinuria is generally regarded as the principal risk factor for deterioration of renal function. The ongoing proteinuria results in progressive renal damage. More than half of the patients with macro albuminuria progress to end-stage renal disease (ESRD) within 10 years.Citation1,Citation2 The glomerular filtration barrier injury is responsible for leakage of high-molecular-weight proteins. It can be secondary to a wide range of medical problems, including various cardiovascular, metabolic and autoimmune diseases, or occur as a heredity disorder as a result of gene mutation. Numerous advances in our understanding of gene variant and renal diseases have been witnessed in the past 10 years. More than 10 genes have been identified as pathogenic forms until now.Citation3–5 Some genetic variants are highly correlated with proteinuria progression and relapse, therefore the identification of a mutation in a patient renders further immunosuppressive treatment unnecessary or disease recurrence in a kidney graft unlikely.Citation6
Podocin, the protein consists of 383 amino acid (AA) and encoded by the NPHS2 gene, is essential for an intact kidney filtration barrier and usually mutated in patients with proteinuria. Its function is currently understood as a scaffold providing the necessary protein lipid micro-environment needed for proper function and signaling of the slit diaphragm protein complex.Citation7–9 NPHS2 knock-out mice developed proteinuria during the antenatal period and died a few days after birth from renal failure caused by massive mesangial sclerosis.Citation10 In previous reports, 20–40% of the familial, autosomal recessive, pediatric steroid-resistant nephrotic syndrome (SRNS) can be ascribed to mutations of the NPHS2 gene. Single-nucleotide polymorphism (SNP) is the main form among these mutations.Citation11,Citation12. In summary, more than 3000 mutations of NPHS2 have been detected in previous study. Nearly 1% of them change codon that codes for different AA or AA deleted. Non-conservative mutation, include missense mutations and non-sense mutation, can render the resulting protein structural changed, but are not always pathogenic. As two or more mutations of these variants usually occur simultaneously, the pathogenic capacity of an oligoallelic form is often ignored in previous studies.Citation13 The evidence of the oligoallelic mutations is weak due to a lack of adequate data and discrepancies between the published investigations. A meta-analysis may clarify the association between the mutations and susceptibility to proteinuria; therefore, we performed meta-analyses to comprehensively investigate the effects of the mutations of podocin on patients with proteinuria in a pooled study.
Methods
Search strategy
Systematic literature searches were conducted in the following electronic databases: PubMed, Embase, Web of Science, Chinese Biomedical Literature (CBM), and the Cochrane Library. The relevant articles were published before 15 January 2014. Non-English publications satisfying the inclusion criteria were translated into English and then assessed. The following search terms were used: (NPHS2 OR Podocin) AND proteinuria AND (polymorphism/polymorphisms OR variant/variants OR mutations/mutation). To identify additional potentially relevant publications, the related references from all the retrieved articles and reviews were manually searched. Only published studies with full-text articles were included in the meta-analysis. All data were entered into the Review Manager 5.0 software (Cochrane Collaboration, Germany) by one author (Lu Lu) and checked by another author (Xiao-ming Sun). Any disagreements were resolved by discussion between the two reviewers and by seeking the opinion of a third reviewer (Yi Yin) when necessary.
Inclusion criteria
Studies were selected based on the following inclusion criteria: (1) at least one AA mutation of podocin was detected in the study. (2) The study was a case–control study or a cohort study of renal diseases with proteinuria. (3) The age of the patients at disease onset was described as either early-onset (onset age <16 years) or all onset ages. (4) The diagnostic criteria of diseases were described in the study and were assessed by professional physicians or biopsies.
Exclusion criteria
Studies were excluded according to the following criteria: (1) a non-human population was mentioned. (2) The study was a case report or a familial investigation. (3) The proteinuria was secondary to anon-renal cause (overflow proteinuria, drug side effect, excessive fluid intake, etc.). (4) The presence of mutations was not assessed in all individuals. (5) All individuals with variants were compound status.
Data extraction and synthesis
Two independent investigators (Lu Lu and Xiao-ming Sun) performed the data extraction using the inclusion and exclusion criteria described above. All discrepancies were resolved by discussion and, if required, participation by the third author (Yi Yin). The following information was extracted from each study: the identical AA mutations, first author’s surname, year of publication, phenotype of diseases, geographical location of the study population, and the number of cases and controls with each AA mutation. Allele frequencies were calculated for the case and control groups from the corresponding genotype distribution. Patients with the compound mutations were removed from the analysis and listed on the tables. The two investigators’ results were compared, and disagreements were resolved by discussion.
Statistical analysis
The statistical analyses were performed using Stata Version10.0 (Stata Corp LP, College Station, TX) software. The included studies were categorized by disease phenotype and the AA mutations. Then, the comparisons of mutations carriers were made between cases and controls by pooling data. Only an AA mutation included by more than one studies was selected to the analyses. The association between the AA mutation and the disease phenotype was determined by calculating the respective odds ratio (OR) with a 95% confidence interval (CI). To validate the meta-analysis results, a sensitivity analysis was performed by the sequential omission of individual studies. Heterogeneity of the effect sizes was evaluated using the Q statistic and the I2 statistic.Citation14 A fixed-effects model was used when the p value was greater than 0.05 and I2 was less than 50%; otherwise, a random effects model was used. The significance of the pooled OR was determined using a Z test, and a p value of less than 0.05 was considered significant. Publication bias was simulated using the Harbord linear regression method. To evaluate our results, two indirect in silico predictors, Mutation Taster (http://www.mutationtaster.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) were used for reference. The above work was completed by two authors and checked by the third author.
Results
Characteristics of the included studies
A total of 236 possibly relevant published articles were identified following the retrieval strategy outlined above. After strict review, 185 of these publications were excluded and 53 were selected for our meta-analysis. shows the selected articles included 64 AA mutations from eight exons of NPHS2 (from 143 comparative trials), of which 59 were studies on proteinuria susceptibility (), eight were studies on steroid response () and five were studies on the recurrence of proteinuria after renal transplantation (). Of the included trials, 60 were conducted in Europe from 13 countries (Belgium, Cyprus, Finland, France, German, Greece, Hungary, Italy, Netherland, Spain, Poland, the UK, and the Czech Republic), 41 were Western Asian and Arabian trials (Egypt, Saudi Arab, and Turkey), 26 were North American trials (the US and Canada), ten were South Asian trials (India and Pakistan), two were from Mexican, and two were form Japan. A total of 45,839 samples (including 22,725 patients and 23,114 healthy controls) were used for the analysis. As only a AA mutation included by more than one studies can be pooled and analyzed, we selected 16 mutations (P20L, Q39X, P118L, P118L, R138X, G140X, V165X, R168H, L169P, V180M, R229Q, A242V, V260E, V268G, A284V, and V290M) on proteinuria susceptibility and two mutations (R229Q and E237Q) on steroid response. As a reference, only synonymous mutations of podocin were found in East Asian patients with proteinuria (163 Chinese, 136 Japanese, and 70 Koreans) and the five relevant studies were not included in the analysis described above.Citation15–21
Figure 1. The included oligoallelic AA mutations on podocin. Human podocin consists of 383 amino acids from 8 exons. A total of 64 AA mutations were analyzed in our study, including 46 AA substitutions and 18 AA deletions. The PHB domain was indicated.
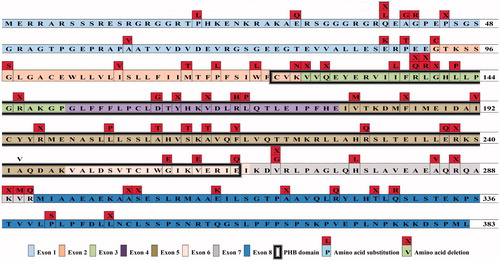
Table 1. The included oligoallelic AA mutations on proteuria susceptibility.
Table 2. Characteristics of the studies that have evaluated the effect of the oligoallelic AA mutations on the steroid response.
Table 3. Characteristics of the studies that have evaluated the effect of the oligoallelic AA mutations on the recurrence of proteinuria after renal transplantation.
Sensitivity analysis
A sensitivity analysis was performed to explore the influence of the trial quality on the magnitude of the effect. We used the METANINF command in Stata to investigate the influence of each individual study on the overall meta-analysis summary estimate.Citation22 Three subgroups were analyzed using a random-effects model, and 24 subgroups were analyzed using a fixed-effects model. For all of the pooled analyses, the significance of the OR was not greatly influenced by omitting any single study. This result demonstrates the stability of the data in our analysis.
Association of the AA mutations with proteinuria susceptibility
A total 99 of the included studies reported 16 AA mutations for the proteinuria patients and controls. In our meta-analysis, we did not find any significant associations between eight AA mutations and proteinuria across the study populations (P20L, Q39X, R138X, V165X, L169P, A242V, V268G, and A284V). The ORs of these AA mutations were not statistically significant (p > 0.05). In the all onset age populations, the ORs that were estimated for proteinuria susceptibility were statistically significant in five mutations: R138Q (OR 10.640, 95% CI 5.937–19.067, p < 0.001), G140X (OR 4.945, 95% CI 1.120–21.829, p = 0.035), R229Q (OR 1.450, 95% CI 1.003–2.097, p = 0.048), V260E (OR 8.466, 95 % CI 1.524–47.038, p = 0.015), and V290M (OR 4.417, 95% CI 1.042–18.718, p = 0.044). In the early-onset (onset age < 16) populations, the ORs that were estimated for proteinuria susceptibility were also statistically significant in five mutations: P118L (OR 5.539, 95% CI 1.336–22.965, p = 0.018), R138Q (OR 11.555, 95% CI 6.112–21.845, p < 0.001), R168H (OR 10.760, 95% CI 1.091–106.16, p = 0.042), V180M (OR 7.997, 95% CI 2.478–25.805, p = 0.001), and V260E (OR 13.359, 95% CI 1.587–112.417, p = 0.017). Three mutations (R140X, R229Q, and V290M) were determined the pathogenic effect in all onset age populations but not in the early-onset populations. The results of Polyphen-2 and Mutation taster were consistent: All selected mutations have been identified to disease causing except for P20L (benign). The details of the results above are listed in .
Table 4. Meta-analysis of the single mutations on NPHS2 and association with proteinuria/steroid response.
Association of the mutations with steroid response and recurrence of proteinuria
Five of the included studies reported two mutations (R229Q and E237Q) allele frequencies for the SRNS patients and steroid-sensitive nephrotic syndrome (SSNS) controls. In our meta-analysis, we did not find any significant associations between the two mutations and steroid resistance across all of the study populations. Furthermore, even the R138Q, the R138X, and the L347X were both mentioned twice in our included studies, only one trial focusing on the L347X set up a control group contained 80 patients without recurrence. In the analysis for the recurrence of proteinuria after renal transplantation, no AA mutation was selected due to lack of control data. The results of Polyphen-2 and Mutation taster show that, except P20L in exon 1, all other selected mutations have been identified to disease causing. The details of the results above are listed in .
Publication bias
Although only three of the subgroups (R138Q, All onset ages group; R138Q, Early-onset group; R229Q, All onset ages group) contained more than 10 studies, Harbord tests were performed to assess the publication bias in this meta-analysis (). The results did not reveal obvious evidence of asymmetry in 24 of the analyses because the p values of the Harbord tests were greater than 0.1. However, the p value of the Harbord test was less than 0.1 for three analyses: the frequency of the P118L on proteinuria susceptibility in early-onset populations (p = 0.013), the frequency of the R138Q on proteinuria susceptibility in all onset ages populations (p = 0.029) and in early-onset populations (p = 0.029). This result suggests that there was a bias in these studies, but the in sufficient number of studies (only four studies included) may have caused the deviation in the bias. The number of studies was too small to conduct the Harbord test in seven of our analysis groups, for which p values could not be calculated.
Discussion
Podocin plays an important role in the slit diaphragm of podocytes. Mutations in this protein gene NPHS2 can cause nephrotic syndrome, such as focal segmental glomerulosclerosis (FSGS) or minimal change disease (MCD), and always related to steroids resistance.Citation7,Citation23,Citation24 The mainstream opinions consider homozygous or compound heterozygous mutations in NPHS2 account for 50% of the cause of proteinuria.Citation13,Citation25 However, the AA mutations were always compound in a patient and trans-associated with each other. The oligoallelic state of each AA mutation was hard to evaluate due to small samples in most experiments. In our study, the pooled results show eight mutations (P118L, R138Q, G140X, R168H, V180M, R229Q, V260E, and V290M) possess the causative effect for proteinuria with oligoallelic state. Of these mutations, three (G140X, R229Q, and V290M) were confirmed their effect in all onset ages subgroup but not statistically significant in early-onset age subgroup. It suggests that the three mutations may be associated with late-onset disease.
We cannot confirm an oligoallelic causal link between the other seven mutations (P20L, Q39X, R138X, V165X, L169P, A242V, and A284V) and the proteinuria. However, only the P20L was assessed as the benign mutation by the mutation taster and the Polyphen2, while the others were identified as disease causing. Perhaps it is because these mutations play as modifiers other than oligoallelic causative factors. For example, A242V and A284V were considered causal in trans-association to R229Q,Citation26–28 and NPHS1 polymorphism may enhance pathogenicity of P118L in podocin.Citation29 Furthermore, the sample size is usually small in control groups. In fact, using data from small sample size in control groups (compared with case groups) have the disadvantage that allele frequencies could be underestimated when merging data.Citation30 With the improvement of population genetics databases in different ethnic groups, using data from whole-genome databases as controls may solve this problem. By the way, only the synonymous mutations, such as A318A and L346L, are found in East Asian patients. It suggests that the mutations in podocin may not be responsible for the proteinuria in East Asian populations.
Among the pathogenic mutations, the R229Q was a hot spot and a controversial issue. This AA exchange with arginine to glutamine substitution is attributed to G to A nucleotide substitution at position 686 in exon 5. It was first reported that R229Q decrease nephrin binding to podocin and cause SRNS by Tsukaguchi et al.Citation31 However, subsequent studies denied its independent pathogenicity in oligoallelic state. Mainstream views generally considered R229Q leads to proteinuria only in association with a pathogenic mutation on the second NPHS2 allele.Citation32,Citation33 In our study, we corrected the selective data by excluding patients with compound R229Q variant, and the pooled results also denied the associations between the R229Q variant and proteinuria in the early-onset study populations, but not in late-onset patients. Although the differences cannot be explained now, we must take into account the sample size and the onset age in these previous studies. The number of participants are usually too little to estimate the causative effect as the low allele frequencies (4–7% in Europeans and 0–1.5% in African and Asian populations), especially for the more rare oligoallelic or homozygous events.Citation34,Citation35. Of the included 21 studies, only three derived positive results. The sample size in case groups is often several times bigger than in healthy control groups, it may lead to an underestimated result. When pooled all data of oligoallelic events, the meta-analysis result do show a statistical significance. On the other hand, some experiments in vitro show that the R229Q exert a deleterious effect on podocin only trans-associated with another pathogenic mutation, but the subjects were adolescent or children (age < 16 years).Citation36 These experiments cannot exclude the hypothesis that the level of podocin is sufficient to maintain glomerular function despite its low expression or properties altered when patients were young. People with R229Q may be pre-disposing to mechanical instability and deterioration of the filtration barrier as a long-term effect. Therefore, we suppose oligoallelic R229Q may possess causative effect, and the age and the compound mutations are both amplification factors. Further studies are warranted to comprehensively investigate the precise relationship.
In general, mutations in the NPHS2 gene are implicated in an autosomal-recessive form of SRNS and recurrence of proteinuria after renal transplantation.Citation6,Citation37 Identification of podocin mutations was considered not only save the patients from unnecessary treatment with cortisol agents but may also assess the prognosis of renal transplant.Citation24,Citation38–40 In our study, however, we found no significant link between steroid resistance and the presence of two including mutations (R229Q and E237Q). This result is consistent with some previous studies.Citation41 Our results show the oligoallelic mutations of podocin cannot currently be considered as potential predictors to the effect of steroid treatment. We also summarized the mutations in regard to the recurrence of proteinuria after renal transplantation. Unfortunately, only one study set up a control group including 80 patients without recurrence of proteinuria. Despite the absence of the meta-analysis, three mutations (R138Q, R138X, and L347X) are mentioned in more than one study and occur with significantly high frequency in case groups. R138X and R138Q are generally considered as the most common mutations in patients with recurrence, which leads to a truncated podocin protein.Citation6 Further studies are warranted to investigate the link between recurrence of proteinuria after renal transplantation and mutations in podocin in large sample sizes.
It should be noted there might be possible limitations in the meta-analysis design. In our raw data, the sample merged familial and sporadic patients together, and the diagnosis was usually based on biopsy other than the presence of proteinuria. It may cause selection bias when pooled. Nonetheless, our meta-analysis tested the pathogenicity of the oligoallelic AA mutations in podocin and suggested the potential causative mutations, and the alleles showing an association with protein susceptibility. The sensitivity and specificity of each causative mutation are pending further testing.
Declaration of interest
We declare that we have no financial or personal relationships with other people or organizations that can inappropriately influence our work; there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, this manuscript. This research is supported by the National Science Foundation of China (No. 81173457 and No. 81001674).
References
- Short versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Lancet. 1988;1(8582):380–383
- Primary nephrotic syndrome in children: Clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. A Report of the International Study of Kidney Disease in Children. Kidney Int. 1981;20(6):765–771
- Fuchshuber A, Jean G, Gribouval O, et al. Mapping a gene (SRN1) to chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a distinct entity of autosomal recessive nephrosis. Hum Mol Genet. 1995;4(11):2155–2158
- Machuca E, Benoit G, Nevo F, et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010;21(7):1209–1217
- Machuca E, Benoit G, Antignac C. Genetics of nephrotic syndrome: Connecting molecular genetics to podocyte physiology. Hum Mol Genet. 2009;18(R2):R185–R194
- Holmberg C, Jalanko H. Congenital nephrotic syndrome and recurrence of proteinuria after renal transplantation. Pediatr Nephrol. 2014;29(12):2309–2317
- Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24(4):349–354
- Caridi G, Bertelli R, Carrea A, et al. Prevalence, genetics, and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulosclerosis. J Am Soc Nephrol. 2001;12(12):2742–2746
- Volker LA, Schurek EM, Rinschen MM, et al. Characterization of a short isoform of the kidney protein podocin in human kidney. BMC Nephrol. 2013;14:102
- Roselli S, Heidet L, Sich M, et al. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol. 2004;24(2):550–560
- McCarthy HJ, Bierzynska A, Wherlock M, et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8(4):637–648
- Buscher AK, Kranz B, Buscher R, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5(11):2075–2084
- Chernin G, Heeringa SF, Gbadegesin R, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23(9):1455–1460
- Zhong C, Zhang Y, Sun J. Acupuncture and acupoint injection for 68 cases of chronic pharyngitis. Zhongguo Zhen Jiu. 2012;32(10):946
- Li J, Ding J, Zhao D, et al. WT1 gene mutations in Chinese children with early onset nephrotic syndrome. Pediatr Res. 2010;68(2):155–158
- Yu Z, Ding J, Huang J, et al. Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children. Nephrol Dial Transpl. 2005;20(5):902–908
- Wu MC, Wu JY, Lee CC, Tsai CH, Tsai FJ. Two novel polymorphisms (c954T > C and c1038A > G) in exon8 of NPHS2 gene identified in Taiwan Chinese. Hum Mutat. 2001;17(3):237
- Kawashima M, Wada K, Ohta H, Terawaki H, Aizawa Y. Association between asymptomatic hyperuricemia and new-onset chronic kidney disease in Japanese male workers: A long-term retrospective cohort study. BMC Nephrol. 2011;12:31
- Sako M, Nakanishi K, Obana M, et al. Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int. 2005;67(4):1248–1255
- Maruyama K, Iijima K, Ikeda M, et al. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr Nephrol. 2003;18(5):412–416
- Cho HY, Lee JH, Choi HJ, et al. WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23(1):63–70
- Wang XW, Ni DF. Gastroesophageal reflux and chronic pharyngitis. Zhonghua Er Bi Yan Hou Ke Za Zhi. 2004;39(1):55–58
- Mollet G, Ratelade J, Boyer O, et al. Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J Am Soc Nephrol. 2009;20(10):2181–2189
- Carraro M, Caridi G, Bruschi M, et al. Serum glomerular permeability activity in patients with podocin mutations (NPHS2) and steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13(7):1946–1952
- Gbadegesin R, Hinkes B, Vlangos C, et al. Mutational analysis of NPHS2 and WT1 in frequently relapsing and steroid-dependent nephrotic syndrome. Pediatr Nephrol. 2007;22(4):509–513
- Tonna SJ, Needham A, Polu K, et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol. 2008;9:13
- Santin S, Tazon-Vega B, Silva I, et al. Clinical value of NPHS2 analysis in early- and adult-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6(2):344–354
- Ardiles LG, Carrasco AE, Carpio JD, Mezzano SA. Late onset of familial nephrotic syndrome associated with a compound heterozygous mutation of the podocin-encoding gene. Nephrology (Carlton). 2005;10(6):553–556
- Dincel N, Mir S, Berdeli A, Bulut IK, Sozeri B. Does NPHS1 polymorphism modulate P118l mutation in NPHS2? Saudi J Kidney Dis Transpl. 2013;24(6):1210–1213
- Kang J, Brant R, Ghali WA. Statistical methods for the meta-analysis of diagnostic tests must take into account the use of surrogate standards. J Clin Epidemiol. 2013;66(5):566.e1–574.e1
- Tsukaguchi H, Sudhakar A, Le TC, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. 2002;110(11):1659–1666
- Kerti A, Csohany R, Wagner L, Javorszky E, Maka E, Tory K. NPHS2 homozygous p.R229Q variant: Potential modifier instead of causal effect in focal segmental glomerulosclerosis. Pediatr Nephrol. 2013;28(10):2061–2064
- Kottgen A, Hsu CC, Coresh J, et al. The association of podocin R229Q polymorphism with increased albuminuria or reduced estimated GFR in a large population-based sample of US adults. Am J Kidney Dis. 2008;52(5):868–875
- Lu L, Wan H, Yin Y, et al. The p.R229Q variant of the NPHS2 (podocin) gene in focal segmental glomerulosclerosis and steroid-resistant nephrotic syndrome: A meta-analysis. Int Urol Nephrol. 2014;46(7):1383–1393
- Santin S, Tazon-Vega B, Silva I, et al. Clinical value of NPHS2 analysis in early- and adult-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6(2):344–354
- Tory K, Menyhard DK, Woerner S, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. 2014;46(3):299–304
- Bouchireb K, Boyer O, Gribouval O, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: A mutation update and the associated phenotypic spectrum. Hum Mutat 2014;35(2):178–186
- Jungraithmayr TC, Hofer K, Cochat P, et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J Am Soc Nephrol. 2011;22(3):579–585
- Furue T, Hattori M, Tsukaguchi H, et al. Clinical features and mutational survey of NPHS2 (podocin) in Japanese children with focal segmental glomerulosclerosis who underwent renal transplantation. Pediatr Transpl. 2008;12(3):341–346
- Ruf RG, Lichtenberger A, Karle SM, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15(3):722–732
- Mishra OP, Kakani N, Singh AK, et al. NPHS2 R229Q polymorphism in steroid resistant nephrotic syndrome: Is it responsive to immunosuppressive therapy? J Trop Pediatr. 2014;60(3):231–237
- Berdeli A, Mir S, Yavascan O, et al. NPHS2 (podicin) mutations in Turkish children with idiopathic nephrotic syndrome. Pediatr Nephrol. 2007;22(12):2031–2040
- Al-Hamed MH, Al-Sabban E, Al-Mojalli H, et al. A molecular genetic analysis of childhood nephrotic syndrome in a cohort of Saudi Arabian families. J Hum Genet. 2013;58(7):480–489
- McKenzie LM, Hendrickson SL, Briggs WA, et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J Am Soc Nephrol. 2007;18(11):2987–2995
- Bakr A, Yehia S, El-Ghannam D, et al. NPHS2 mutations. Indian J Pediatr. 2008;75(2):135–138
- Lahdenkari AT, Suvanto M, Kajantie E, Koskimies O, Kestila M, Jalanko H. Clinical features and outcome of childhood minimal change nephrotic syndrome: Is genetics involved? Pediatr Nephrol. 2005;20(8):1073–1080
- Jaffer A, Unnisa W, Raju D, Jahan P. NPHS2 mutation analysis and primary nephrotic syndrome in South Indians. Nephrology (Carlton). 2014;19(7):398–403
- Reiterova J, Safrankova H, Obeidova L, et al. Mutational analysis of the NPHS2 gene in Czech patients with idiopathic nephrotic syndrome. Folia Biol (Praha). 2012;58(2):64–68
- Lipska BS, Iatropoulos P, Maranta R, et al. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney Int. 2013;84(1):206–213
- Abid A, Khaliq S, Shahid S, et al. A spectrum of novel NPHS1 and NPHS2 gene mutations in pediatric nephrotic syndrome patients from Pakistan. Gene. 2012;502(2):133–137
- Caridi G, Bertelli R, Scolari F, Sanna-Cherchi S, Di Duca M, Ghiggeri GM. Podocin mutations in sporadic focal-segmental glomerulosclerosis occurring in adulthood. Kidney Int. 2003;64(1):365
- Carrasco-Miranda JS, Garcia-Alvarez R, Sotelo-Mundo RR, Valenzuela O, Islas-Osuna MA, Sotelo-Cruz N. Mutations in NPHS2 (podocin) in Mexican children with nephrotic syndrome who respond to standard steroid treatment. Genet Mol Res. 2013;12(2):2102–2107
- Koziell A, Grech V, Hussain S, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet. 2002;11(4):379–388
- Chernin G, Heeringa SF, Vega-Warner V, Schoeb DS, Nurnberg P, Hildebrandt F. Adequate use of allele frequencies in Hispanics – A problem elucidated in nephrotic syndrome. Pediatr Nephrol. 2010;25(2):261–266
- Ozaltin F, Heeringa S, Poyraz CE, et al. Eye involvement in children with primary focal segmental glomerulosclerosis. Pediatr Nephrol. 2008;23(3):421–427
- Ruf RG, Lichtenberger A, Karle SM, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15(3):722–732
- Megremis S, Mitsioni A, Mitsioni AG, et al. Nucleotide variations in the NPHS2 gene in Greek children with steroid-resistant nephrotic syndrome. Genet Test Mol Biomarkers. 2009;13(2):249–256
- Caridi G, Bertelli R, Di Duca M, et al. Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol. 2003;14(5):1278–1286
- Lowik M, Levtchenko E, Westra D, et al. Bigenic heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2008;23(10):3146–3151
- Santin S, Bullich G, Tazon-Vega B, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6(5):1139–1148
- Karle SM, Uetz B, Ronner V, Glaeser L, Hildebrandt F, Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13(2):388–393
- Kerti A, Csohany R, Szabo A, et al. NPHS2 p.V290M mutation in late-onset steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2013;28(5):751–757
- Caridi G, Gigante M, Ravani P, et al. Clinical features and long-term outcome of nephrotic syndrome associated with heterozygous NPHS1 and NPHS2 mutations. Clin J Am Soc Nephrol. 2009;4(6):1065–1072
- Laurin LP, Lu M, Mottl AK, Blyth ER, Poulton CJ, Weck KE. Podocyte-associated gene mutation screening in a heterogeneous cohort of patients with sporadic focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2014;29:2062–2069
- Lipska BS, Balasz-Chmielewska I, Morzuch L, et al. Mutational analysis in podocin-associated hereditary nephrotic syndrome in Polish patients: Founder effect in the Kashubian population. J Appl Genet. 2013;54(3):327–333
- Ismaili K, Pawtowski A, Boyer O, Wissing KM, Janssen F, Hall M. Genetic forms of nephrotic syndrome: A single-center experience in Brussels. Pediatr Nephrol. 2009;24(2):287–294
- Hinkes B, Vlangos C, Heeringa S, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2008;19(2):365–371
- Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007;119(4):e907–e919
- Voskarides K, Arsali M, Athanasiou Y, Elia A, Pierides A, Deltas C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol. 2012;27(4):675–679
- Vasudevan A, Siji A, Raghavendra A, Sridhar TS, Phadke KD. NPHS2 mutations in Indian children with sporadic early steroid resistant nephrotic syndrome. Indian Pediatr. 2012;49(3):231–233
- Buscher AK, Konrad M, Nagel M, et al. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol. 2012;78(1):47–53
- Machuca E, Hummel A, Nevo F, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75(7):727–735
- Kyrieleis HA, Lowik MM, Pronk I, et al. Long-term outcome of biopsy-proven, frequently relapsing minimal-change nephrotic syndrome in children. Clin J Am Soc Nephrol. 2009;4(10):1593–1600
- Aucella F, De Bonis P, Gatta G, et al. Molecular analysis of NPHS2 and ACTN4 genes in a series of 33 Italian patients affected by adult-onset nonfamilial focal segmental glomerulosclerosis. Nephron Clin Pract. 2005;99(2):c31–c36
- He N, Zahirieh A, Mei Y, et al. Recessive NPHS2 (Podocin) mutations are rare in adult-onset idiopathic focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2007;2(1):31–37
- Weber S, Gribouval O, Esquivel EL, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 2004;66(2):571–579
- Bertelli R, Ginevri F, Caridi G, et al. Recurrence of focal segmental glomerulosclerosis after renal transplantation in patients with mutations of podocin. Am J Kidney Dis. 2003;41(6):1314–1321