
Abstract
Objective: The objective of this study was to measure the 3′-untranslated region (3′-UTR) polymorphism lengths in peripheral blood mononuclear cells (PBMCs) from uremia patients.
Method: We sequenced the alternative polyadenylation sites in the 3′-UTR of PBMCs from 10 uremic patients and 10 healthy people to detect different gene expression levels between uremia patients and healthy controls. Quantitative reverse transcription polymerase chain reaction was used as validation. Result: Compared with the healthy control group, 691 genes in uremic patients had significantly different 3′-UTR lengths. Of these genes, 475 genes showed shortened 3′-UTRs, and the 3′-UTRs of 216 genes were lengthened. The verification results matched the original sequencing results. Conclusion: There were significant differences in 3′-UTR lengths between uremic patients and healthy controls, and analysis of the differential genes may contribute to the understanding of uremia pathogenesis.
Introduction
During chronic renal failure, the function of the kidney progressively and irreversibly decreases due to various renal diseases until it presents a series of symptoms and a clinical syndrome indicative of metabolic disorder. The final phase of chronic renal failure is called uremia. Therefore, uremia is not a single disease but is considered the clinical syndromes caused by terminal renal diseases. Uremia was once considered an incurable disease due to its high morbidity rate and mortality rate. Although dialysis can reduce the toxin concentration in a patient’s body, this therapy does not address the “renal cell decay” problem, which is the basic problem of uremia.
Several parts of mRNA are not translated into protein, including the 5′cap, 5′-untranslated region (5′-UTR), 3′-UTR, and poly (A) tail. The 3′-UTR is a special part of the mRNA that is located in the 3′-end of the coding region, and it immediately follows the translation termination codon. 3′-UTRs affect post-transcriptional gene expression, which is regulated by the cooperative interaction of a cis-acting element in the 3′-UTR and a trans-acting element.Citation1
3′-UTRs can affect translation efficiency, subcellular localization and stability of mRNA.Citation2 The 3′-UTR includes two types of binding sites that regulated by protein or miRNA.Citation3 By binding to certain sites in the 3′-UTR, miRNAs can suppress the translation or directly cause transcripts to degrade, thus decreasing gene expression.Citation4 There are also areas within the 3′-UTR that can bind to repressor proteins to suppress the expression of mRNA.Citation5 Many 3′-UTRs include the ARE element that is rich in AUs, which can affect the start of translation.Citation6 In addition to the sequence, the physical properties of the 3′-UTR, including its length and secondary structure, also affect translation control. These gene regulatory mechanisms ensure that genes are expressed at the proper time and place.
The functions of different genes with 3′-UTR length polymorphisms vary. For example, these polymorphisms may affect the functions of the cell cycle, apoptosis, or metabolism.Citation7 However, there is still a lack of systemic studies about the mechanism of 3′-UTRs controlling gene expression and the progress of uremia. Discussing and analyzing the impact of 3′-UTR length polymorphisms on uremia is a new approach to observe the pathological process of this disease.
Materials and methods
Clinical diagnosis
Ten chronic renal insufficiency patients with uremia (5 females and 5 males; 54.2 ± 15.0 years old) who were treated at the People’s Liberation Army 181 St Hospital from June 2013 to October 2013 were selected for this study. Ten healthy people were selected as normal controls (5 females and 5 males; 42.1 ± 5.5 years old). According to opinions of the National Glomerular Disease Symposium (1992), chronic renal insufficiency uremia is diagnosed when the serum creatinine reaches 707 μmol/L or higher. The diagnosis also involves the presence of the primary disease, chronic glomerulonephritis, as well as the clinical symptoms of uremia. The healthy control group was not associated with autoimmune diseases, recent infection or kidney disease.
Sample information
Two milliliters of peripheral blood was drawn from the 10 patients and 10 healthy controls. Mononuclear cells were isolated by Ficoll density gradient centrifugation. Lymphocyte separation medium was CEDARLANE Lympholyte-H (Cedarlane; Burlington, ON, Canada) and the procedure was performed according to the product instructions. Mononuclear cells were collected in 1.5 mL centrifugal tubes. One milliliter of TRIZOL (Cat. no. 405-02; Tiangen, Beijing, China) was added to lyse the cells and extract total RNA, and the resulting RNA was stored at −80 °C.
RNA extraction and quality inspection
RNA was extracted with the total RNA extraction reagent according to the RNA extraction SOP.Citation8 Ten tubes equally mixed were used as one set in the experiment. A micro UV spectrophotometer (NanoDrop ND-1000, Wilmington, DE) was used to inspect the total amount of RNA in the samples (>3 μg; 1 μg was used for quality inspection; 0.5 μg was used for 3′-UTR library preparation; 0.5 μg was used for experiment backup and 1 μg was used for experiment verification) with OD260/280 values between 1.8 and 2.2. The integrity of the samples was inspected by agarose gel electrophoresis.
3′-UTR library construction and SAPAS
Sequencing of alternative poly(A) sites (SAPAS) is a technology based on the Illumina-Solexa sequencing platform for 3′-UTR-specific detection. The SAPAS process includes mRNA fragmentation by heating, single-strand cDNA synthesis and polymerase chain reaction (PCR) amplification. Specific details for the SAPAS library preparation have been previously published by Yonggui Fu.Citation9 The StepOne™ Real-Time PCR System (Applied Biosystems®, Foster City, CA) was used to inspect the library concentration, which was >1 ng/μL. PCR is a procedure employed to enrich library fragments in platforms of second-generation high-throughput sequencing. A sequence linker comprises a sequencing primer binding site. Only the DNA sequence with complete joint sequencing can be sequenced. Increased numbers of amplification cycles result in better enrichment. However, PCR introduces problems, such as base mutations and redundant fragments, and PCR has different efficiencies resulting from different templates. Therefore, the amplification cycle number is limited to 5–15, and it is 17 in our study. An Agilent Bioanalyzer 2100 (Palo Alto, CA) was used to inspect the main peak of library fragments (disturbance will influence the effectiveness of sequence data).
Real-time RT-PCR verification
Two genes with extreme 3′-UTR length differences between uremia patients and the normal controls were selected. The region upstream of the supersites [the proximal and distal poly(A) sites] was targeted for quantitative reverse transcription PCR (qRT-PCR) (Bio-Rad, MiniOpticon, Hercules, CA).
Results
RNA extraction and quality inspection
The RNA was inspected by an UV spectrophotometer (NanoDrop), Agilent 2100 Bioanalyzer and agarose gel electrophoresis, and it was presented to be comparatively complete. All the indexes of RNA sample quality met the quality inspection standard. The total RNA was >17 μg, and the OD260/280 was >1.97. Thus, a 3′-UTR library was able to be constructed.
Library construction and library quality inspection
The total library of the experimental group was 59.85 ng, and that of the control group was 75.00 ng. The Agilent Bioanalyzer 2100 showed that all the library fragments were ∼180–680 bp (average size of the experimental group was 365 bp, and the average size of the control group was 391 bp), and the concentration of the library was qualified. The results met expectation, and the sequencing was then started.
3′-UTR sequencing and bioinformatics analysis
The raw sequencing data were filtered and trimmed. After filtering internal priming reads, the clean reads of the control and experimental groups were 23,320,710 and 22,843,116, respectively. Thus, the reads met the quality requirements that single-terminal sequencing effective reads should be >10,000,000/sample. Most reads were mapped to the human genome (version: UCSC, hg19). Both the control group and the experimental group had >87% of the reads mapped to the human genome. At least 63.2% of the reads were uniquely mapped to the human genome. Of these reads, the majority uniquely mapped to (>56.1%) the nuclear genome. After passing the internal priming filter, there were 9.6 and 12.4 million (M) reads for subsequent detection of transcription splice sites for the control and experimental groups, respectively. The reads mapped to the mitochondrial genome were no more than 8%, indicating that this section could not be considered in the subsequent analysis.
For protein-coding mRNA, poly(A) sites determine the termination of the 3′-UTR. One gene often has more than one poly(A) site (alternative polyadenylation; APA), thereby resulting in the same gene having transcripts with different 3′-UTRs. Due to the heterogeneity of the cleavage sites at poly(A) sites, we considered cleavage clusters with more than one read as poly(A) sites.Citation10 We identified 139,854 poly(A) sites in the experimental peripheral blood mononuclear cells (PBMCs) and 224,262 poly(A) sites in the control PBMCs. Moreover, 14,764 and 14,111 genes had more than one tandem APA site in the control and experimental groups, respectively. The majority of reads fell within 24 nt of the known poly(A) sites, and a small number of reads (<3%) fell into the 3′-UTR of the UCSC gene model. However, we found <20% of poly(A) sites in the UCSC and Tian’s databases, thus suggesting that the SAPAS method identified the new poly(A) sites more effectively despite their low expression levels.
Genes containing two or more tandem poly(A) sites are called tandem 3′-UTR genes. We compared the tandem 3′-UTR lengths of the uremia patient PBMCs to those of the normal controls. For tandem 3′-UTR genes expressed in both samples, we performed a linear trend alternative to the independence test in the analysis of 3′-UTR length polymorphisms.Citation11 The proportion of the use of different APA sites may change, and this phenomenon is defined as APA site switching, which causes various 3′-UTR lengths.
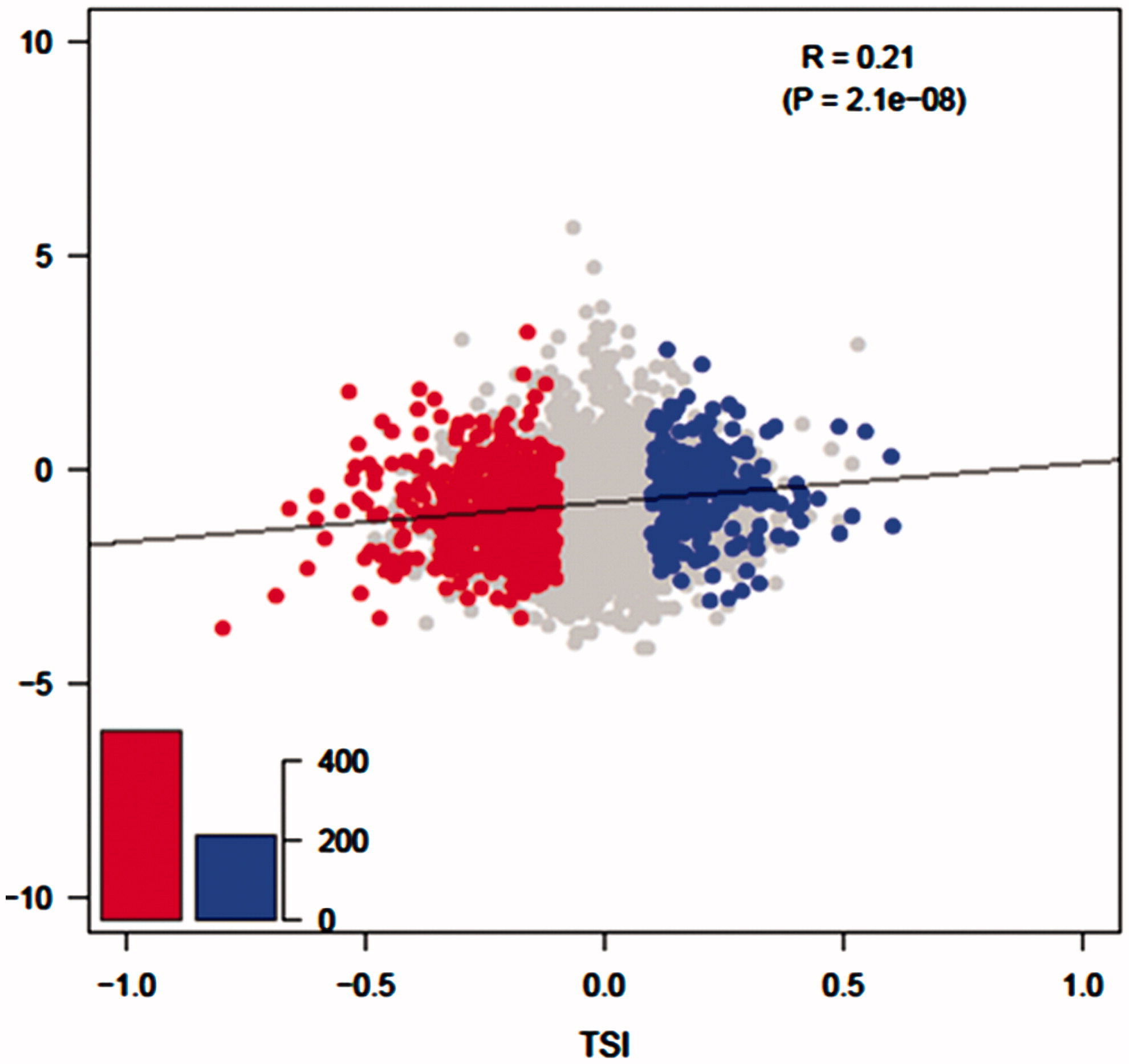
We denoted the normal control as 1 and the uremia group as 2. A positive Pearson correlation r indicates that sample 2 harbors longer tandem 3′-UTRs than sample 1, and a negative Pearson correlation r indicates that sample 2 harbors shorter tandem 3′-UTRs than sample 1. Here, we defined the Pearson correlation r as tandem APA sites switch index (TSI). We found 691 genes [false discovery rate (FDR) < 0.01] with a significant difference in the tandem 3′-UTR length between samples 1 and 2, and the TSIs of 69% of these genes were negative ().
Figure 1. Uremic group vs control group 3′-UTR length polymorphism. The horizontal axis represents TSI. Genes have significant change in tandem 3′-UTRs lengths are (FDR < 0.01) marked out as points: genes using a lengthened 3′-UTR are on the right (TSI > 0), and genes using shortened 3′-UTR are on the left (TSI < 0). Y-axis represents the logarithm of the expression level of genes from the uremia sample relative to the normal sample.
Analysis of gene function
We performed functional annotation of these genes using the DAVID Bioinformatics Resources 6.7. The results of Gene Ontology (GO) analyzes indicated that 587 GO terms were involved with 3′-UTR shortened genes and that 264 GO terms were involved with 3′-UTR lengthened genes. The following 17 KEGG pathways were enriched in the genes with shortened 3′-UTRs in the uremia PBMCs: insulin signaling pathway, long-term potentiation, spliceosome, B cell receptor signaling pathway, Notch signaling pathway, apoptosis, renal cell carcinoma, regulation of actin cytoskeleton, Toll-like receptor signaling pathway, T cell receptor signaling pathway, vascular smooth muscle contraction, and chemokine signaling pathway. In contrast, only two KEGG pathways, namely ubiquitin mediated proteolysis and protein export, were enriched in the genes with lengthened 3′-UTRs in the uremia PBMCs. We identified 792 genes that were up-regulated by at least 3-fold and 252 genes that were down-regulated by at least 3-fold. In the up-regulated genes, 765 GO terms were enriched, and 179 GO terms were enriched in down-regulated genes. Up-regulated genes in uremia samples were involved in lysosomes, antigen processing, antigen presentation, hematopoietic cell lineage and protein export. Down-regulated genes were involved in the ribosome, spliceosome and natural killer cell-mediated cytotoxicity. Despite several 3′-UTR lengthened genes being up-regulated and several 3′-UTR shortened genes being down-regulated, we did not observe a positive correlation between 3′-UTR length and gene expression level. Some 3′-UTR lengthened genes and 3′-UTR shortened genes were classified with the same GO terms. For example, 95 genes with longer 3′-UTRs in the uremia sample were associated with cellular macromolecule metabolic processes, and 198 genes with shorter 3′-UTRs in the uremia sample were also associated with cellular macromolecule metabolic processes. These data demonstrated the complexity of the APA site switch and the resulting complex relations between 3′-UTR length polymorphisms and gene function.
Real-time quantitative PCR test
We selected two genes to use for validation (SFRS2B and UBL4A). We tested whether significant 3′-UTR length differences existed between the two samples. These two genes were consistent with the sequencing data. In the uremia sample, SFRS2B had a shortened 3′-UTR, and UBL4A had a lengthened 3′-UTR.
Discussion
Uremia is the final result of glomerular fibrosis, which may be affected by APA control.Citation12 In this study, the 3′-UTR lengths and mRNA expression levels of uremia patients and healthy people were compared. Hundreds of genes were determined to be involved in uremia. Uremia is affected by a variety of factors, and its pathogenesis is complex. These study results were significant in revealing the primary disease and the complicated pathogenesis in uremic patients.
The 3′-UTR plays a role in the regulation of gene expression that is mediated by miRNAs or specific proteins.Citation13 Almost all of the known human mRNA transcripts have more than one miRNA target site.Citation14 Changes in 3′-UTR length cause the loss or gain of these target sites.Citation15 APA commonly exists in the human genome. Genes can produce 3′-UTRs with various lengths by APA, which may imply a new epigenetic regulation of uremia.Citation16 Changes in 3′-UTR lengths mediated by APA sites are a coordinated mechanism for regulating the expression of genes in various physiological and pathological processes, such as T cell activation, cellular transformation, and immune responses.Citation17 In our study, the genes that were identified to undergo APA site-switching events included genes involved in ubiquitin-mediated proteolysis, B cell receptor signaling pathway, apoptosis and many other functions. The conversion of normal cells into cells with failed function is a progressive and dynamic process that is driven by changes of some genes under various environmental forces.
Our study did not directly focus on the issue of glomerular fibrosis, but it investigated the role of APA in gene expression regulation. Our studies indicated that there are significant alterations in 3′-UTR length in uremia patients. These significant 3′-UTR shortened/Lengthened genes may help to explain the primary diseases and the complications in uremia patients. Studying 3′-UTR length polymorphisms in mononuclear cells in uremia peripheral blood at a genomic level can provide potential biomarkers for uremia with regard to early diagnosis, prognosis, targeted therapy and individualized treatment. This type of study can also provide new ideas and potential approaches for the treatment of this disease.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article. This work was supported by Research Funds for Guilin Technology Plan (20130121-6), Guangxi Natural Science Foundation of China (No. 2014GXNSFAA118179) and Guangxi Key Laboratory Construction Projects Planned Mission Statement (14–045-41).
References
- Díaz-Muñoz MD, Bell SE, Turner M. Deletion of AU-rich elements within the Bcl2 3′UTR reduces protein expression and B cell survival in vivo. PloS One. 2015;10:e0116899.
- Deling L, Ying L, Yi S. The post-transcriptional regulation of the DNA damage response. Hereditas. 2014;36:309–315.
- Vislovukh A, Vargas TR, Polesskaya A, et al. Role of 3′UTR translational control in cancer. World J Biol Chem. 2014;5:e51.
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524.
- Miles WO, Dyson NJ. Pumilio and nanos RNA-binding proteins counterbalance the transcriptional consequences of RB1 inactivation. Mol Cell Oncol. 2014;1:e968074.
- Vlasova St, Louis I Bohjanen PR. Post-transcriptional regulation of cytokine signaling by AU-rich and GU-rich elements. J Interferon Cytokine Res. 2014;34:233–241.
- Chang H, Lim J, Ha M, et al. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol Cell. 2014;53:1044–1052.
- Ugalde-Olano A, Egia A, Fernández-Ruiz S, et al. Methodological aspects of the molecular and histological study of prostate cancer: Focus on PTEN. Methods. 2015;77:25–30.
- Fu Y, Sun Y, Li Y, et al. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21:741–747.
- Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212.
- Li Y, Sun Y, Fu Y, et al. Dynamic landscape of tandem 3′ UTRs during zebrafish development. Genome Res. 2012;22:1899–1906.
- Taotao T, Linli L, Bicheng L. MicroRNAs and renal fibrosis. J Southeast Univ (Medical Sci Edi). 2014;33:788–793.
- Lutz CS, Moreira A. Alternative mRNA polyadenylation in eukaryotes: An effective regulator of gene expression. Wiley Interdiscip Rev RNA. 2011;2:22–31.
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
- Sandberg R, Neilson JR, Sarma A, et al. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647.
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463.
- Tian P, Sun Y, Li Y, et al. A global analysis of tandem 3′UTRs in eosinophilic chronic rhinosinusitis with nasal polyps. PLoS One. 2012;7:e48997.