
Abstract
We determined the effect of indoxyl sulfate (IS) on Pit-1 expression and the role of Pit-1 in IS-induced osteoblastic differentiation and calcification of vascular smooth muscle cells (VSMCs). To assess osteoblastic differentiation and Pit-1 expression, VSMCs were incubated with various concentrations of IS for different durations. Phosphonoformic acid (PFA), a competitive inhibitor of Pit-1, was used to verify the role of Pit-1. Western blot analysis and quantitative real-time polymerase chain reaction (PCR) were performed to assess Pit-1 protein and mRNA levels, respectively. To evaluate calcification, calcium content was measured. After IS treatment, we observed osteoblastic differentiation and calcification of VSMCs and up-regulation of Pit-1 expression. Moreover, the effect of IS on osteoblastic differentiation and Pit-1 expression was partly dose- and time-dependent. PFA abrogated the IS-induced osteoblastic differentiation and calcification of VSMCs to a certain extent. The c-Jun N-terminal kinase (JNK) pathway was activated after treatment with IS, whereas inhibition of the JNK pathway partially attenuated the effect of IS on both the stimulation of Pit-1 expression and calcium deposition. Our study is the first to demonstrate that IS promotes Pit-1 expression in part by activation of the JNK pathway that is involved in the mechanism of IS-induced osteoblastic differentiation and matrix mineralization.
Introduction
The morbidity and mortality of cardiovascular diseases (CVD) are higher in patients with chronic kidney disease (CKD) than in patients without CKD. In fact, CVD accounts for the premature death of more than 50% of patients undergoing regular dialysis.Citation1 Vascular calcification is an independent risk factor for the development of CVD and a prognostic indicator of end-stage renal disease (ESRD).Citation2 Vascular calcification affects both vascular intimal and media layers, but the underlying mechanism remains poorly understood. Despite the fact that the calcification mechanism is not fully understood, elevated serum phosphorus (Pi) has emerged as a key risk factor for vascular calcification in patients with CKD and the general population.Citation3–5 Indeed, Pi is an important regulator of vascular calcification and a risk factor for cardiovascular mortality in dialysis patients. It has been widely established that high extracellular Pi induces matrix calcification of vascular smooth muscle cells (VSMCs) in vitro.Citation6 In particular, elevated Pi has been linked to several highly regulated processes in the vessel wall, which promote VSMC calcification. These processes include VSMC osteogenic differentiation, apoptosis, and matrix degradation.Citation7,Citation8 The phenotype change is particularly striking because the VSMCs cease to express smooth muscle markers and begin to express bone-forming genes.
Pi transport via type-III sodium (Na)-Pi cotransporters is a crucial step in calcification.Citation9 Type-III Na-Pi cotransporters (known as the SLC20 family), PiT-1 and PiT-2, are ubiquitously expressed throughout the body and are the major Pi transport proteins found in VSMCs.Citation10,Citation11 In human VSMCs, PiT-1 is the predominant Pi transporter with 8-fold higher mRNA expression than PiT-2.Citation12 Inhibition of Pit-1 blocks both Pi-induced phenotypic transition and calcification of VSMCs.Citation13,Citation14 This finding suggests that elevated Pi via a Pit-1-mediated mechanism has direct pro-mineralizing effects on VSMCs. Therefore, a regulator of Pit-1 expression may affect calcification.
In patients with CKD, calcification of the vascular media layers, a feature of osteoblastic differentiation of VSMCs, is pervasive. In addition to the traditional cardiovascular risk factors, hyperphosphatemia, calcium overload, increased oxidized low-density lipoprotein cholesterol, uremic toxins, increased oxidative stress, hyperhomocysteinemia, hemodynamic overload, and dialysate-related factors might play a role in vascular calcification.Citation15 Indoxyl sulfate(IS), a circulating uremic toxin, is involved in the progression of not only CKD but also CVD.Citation16 In fact, the serum IS concentration may predict the initiation of CVD in patients with CKD.Citation17 IS is a metabolite of tryptophan derived from dietary protein and synthesized in the liver from indole that is produced by intestinal flora including Escherichia coli. It is normally excreted into urine. As renal functions deteriorate, IS accumulates because of reduced renal clearance. Current conventional hemodialysis cannot remove IS because 90% of IS is bound to albumin, and IS–albumin complexes are too large to pass through the pores of dialysis membranes. In addition to its nephrotoxicity, IS is a cardiovascular toxin. It has been reported that IS induces endotheliocyte dysfunction,Citation18,Citation19 arteriosclerosis,Citation20 vascular calcification,Citation21 and myocardial fibrosis,Citation22 resulting in cardiovascular system impairment. Although IS induces expression of osteoblast-specific proteins in VSMCsCitation23 and vascular calcification in hypertensive rats,Citation21 probably via enhanced oxidative stress, the mechanism by which IS leads to calcification in ESRD remains poorly understood. Because regulation of Pit-1 expression affects calcification, whether IS promotes osteoblastic differentiation and calcification by enhancing Pit-1expression is unknown.
Methods and materials
Cell culture
Primary human umbilical vein smooth muscle cells (HUVSMCs) were purchased from ScienCell Research Laboratories (San Diego, CA). HUVSMCs were cultured in Smooth Muscle Cell Medium containing 2% fetal bovine serum (ScienCell). The medium was changed every two days. Cells were treated with various concentrations of IS (100, 250, 500, and 1000 μM, 13,875; Sigma, St. Louis, MO) for different durations (12, 24, 48, and 72 h for protein analysis, and 6, 12, 24, 3, 6 and 48 h for mRNA analysis). When indicated, phosphonoformic acid (PFA, P6801; Sigma) or SP600125 (SP, sc-200635; Santa Cruz Biotechnology, Santa Cruz, CA) was included in the treatments. Actively growing VSMCs of the third to fifth passage were used for experiments.
Measurement of calcium content
VSMCs in 12-well plates were cultured in medium with or without 500 μM IS for 10 or 20 days. Cells were washed with phosphate-buffered saline (PBS) and decalcified with 0.6 N HCl for 24 h. Calcium content was determined using the O-cresolphthalein complex one method. After decalcification, the cells were solubilized in 0.1 N NaOH/0.1% SDS. Total protein content was measured with a bicinchoninnic acid (BCA) protein assay kit (Sigma). The calcium content of the cells was normalized to the protein concentration.
To investigate the effect of PFA and SP on calcium content, the method was the same as described previously.
Alizarin red S staining
For Alizarin red S staining, VSMCs in 12-well plates were cultured in medium with or without 500 μM IS for 14 days. The cells were then fixed in 95% ethanol for 30 min at room temperature and stained with 40 mM Alizarin Red S for 10 min. Then, the cells were washed with PBS to eliminate nonspecific staining. Stained matrix was examined under an Eclipse E600 epifluorescence microscope equipped with a digital camera (Nikon, Melville, NY).
Western blot analysis
Cells were lysed in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2%SDS, 10% glycerol, 50 mM DTT, and 0.1% bromophenol blue). Protein concentrations were determined using the BCA protein assay kit. Cell lysates were mixed with an equal amount of 2 × SDS loading buffer (125 mM Tris-HCl, 4% SDS, 20% glycerol, 100 mM DTT, and 0.2% bromophenol blue). Samples were heated at 100 °C for 5–10 min before loading and then separated on precast 10% or 5% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA). The proteins were electro transferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL) in transfer buffer (48 mM Tris-HCl, 39 mM glycine, 0.037% SDS, and 20% methanol) at 4 °C for 1 h. Nonspecific binding to the membrane was blocked for 1 h at room temperature with 5% nonfat milk powder in tris-buffered saline (TBS) buffer (20 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween 20). The membranes were then incubated for 16 h at 4 °C with various primary antibodies in blocking buffer at the dilutions specified by the manufacturers. Primary antibodies were as follows: antibone morphogenetic protein-2 (BMP-2, ab183729; Abcam, Cambridge, UK), antiosteoprotegerin (OPG, ab73400; Abcam), and anti-Pit-1 (ab177147; Abcam). After extensive washing with TBS buffer, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad) in 1% nonfat milk powder in TBS buffer for 1 h at room temperature. The membranes were then washed with TBS buffer, and the signals were visualized using the enhanced chemiluminescence system (ECL; Amersham).
Quantitative real-time PCR
Total RNA was isolated with Trizol reagent (Invitrogen, Carlsbad, CA) and methyl trichloride according to the manufacturer’s instructions. The RNA concentration and quality were checked by spectrophotometry. First-strand cDNA was synthesized from 2 μg RNA by reverse transcription using AMV-RT (Promega, Madison, WI) and random primers at 42 °C for 30 min. Quantitative RT-PCR was performed on the ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA). The 25 μL reaction mixture contained 12.5 μL of 2 × SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 5 μL diluted cDNA (1:10), and 0.5 μM sense and antisense primers. The primers were designed using Primer Express software v.2.0 (Applied Biosystems, Foster City, CA) and their sequences were: human Pit-1 forward 5′-GCCAAAGTGAGCGAAACCATCC-3′ and reverse 5′-CCACACAGCAGAACCAAACATAGC-3′; ratPit-1forward 5′-TGGTGTTGGCATTTGCATGG-3′ and reverse 5′-CGACCACAGTGAAGGCAGAT-3′. Amplification was carried out under the following conditions: initial denaturation for 10 min at 95 °C, followed by cycles of denaturation for 10 s at 95 °C, annealing for 30 s at the optimal temperature for each primer pair, and extension for 30 s at 72 °C. The mRNA levels of target genes were calculated after normalization to glyceraldehyde-3-phosphate dehydrogenase mRNA.
Statistical analysis
All data are expressed as the mean ± standard deviation (SD). Statistical analysis was performed using SPSS version 17.0 software (SPSS Inc., Chicago, IL). Comparisons between groups were made using one-way analysis of variance (ANOVA) followed by the Student–Newman–Keuls test. p < 0.05 was considered statistically significant.
Results
IS affects osteoblastic differentiation and calcification of VSMCs
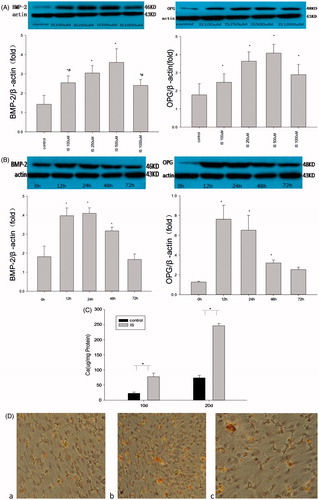
To clarify whether IS induces osteoblastic differentiation of VSMCs, we examined the effect of IS on the expression of osteoblast-specific proteins. The effect of IS on BMP-2 and OPG expression in VSMCs was examined by 24 h of treatment with various concentrations of IS (100, 250, 500, and 1000 μM) and treatment with 500 μM IS for various durations (12, 24, 48, and 72 h). Western blot analysis demonstrated increases in the BMP-2 and OPG expression of VSMCs after IS treatment in a dose- and time-dependent manner (). Next, we analyzed the calcification of VSMCs. First, to examine the calcium content, the VSMCs were treated with 500 μM IS for 10 and 20 days. The results indicated that IS-promoted calcium deposition compared with the control group (). Next, we performed Alizarin Red S staining to investigate calcium deposition. We found that calcium deposits were increased significantly by treatment of the cells with IS ().
Figure 1. Effects of IS on osteoblast differentiation and calcification of VSMCs. (A) VSMCs were incubated with various concentrations of IS for 24 h. (B) VSMCs were incubated with 500 μM IS for different durations. Top panels: representative western blots of BMP-2 and OPG. Graphs: quantification of the expression. (C) VSMCs were incubated with or without 500 μM IS for 10 or 20 days. (D) Alizarin Red S staining. The arrows point to calcium nodes. a: VSMCs were incubated for 14days. b: VSMCs were incubated for 14 days with 500 μM IS. c: VSMCs were incubated with IS and phosphonoformic acid (PFA). Results are presented as percentage of control values and are mean ± SD of three independent experiments. *p < 0.05, compared with the control group. #p < 0.05, compared with the 500 μM IS-stimulated group.
IS affects Pit-1 expression in VSMCs
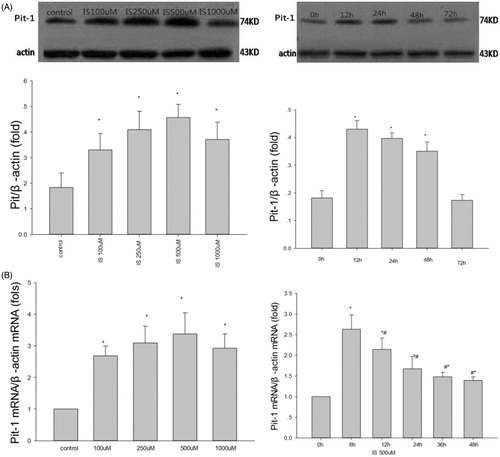
To verify whether IS-induced osteoblastic differentiation of VSMCs is related to Pit-1, the effect of IS on Pit-1 expression was determined at both mRNA and protein levels. The effect of IS on Pit-1 protein expression in VSMCs was examined by 24 h of treatment with various concentrations of IS (100, 250, 500, and 1000 μM) and treatment with 500 μM IS for various durations (12, 24, 48, and 72 h). To examine Pit-1 mRNA levels, VSMCs were treated with the above-mentioned IS concentrations for 6 h or with 500 μM IS for 6, 12, 24, 36, and 48 h. Both western blot analysis () and RT-PCR () demonstrated that IS increased Pit-1 expression in VSMCs at both protein and mRNA levels. Moreover, the peak effect on protein expression was observed at 500 μM IS stimulation for 12 h.
Figure 2. Effect of IS on Pit-1 expression in VSMCs. (A) IS-induced Pit-1 protein expression in a dose- (left) and time-dependent (right)manner. (B) IS-induced Pit-1 mRNA expression in a time-dependent manner (right). In the dose relationship (left). Results are presented as percentage of control values and are mean ± SD of three independent experiments. *p < 0.05, compared with the control group. #p < 0.05, compared with the IS 6-h-stimulated group.
The role of Pit-1 in IS-induced osteoblastic differentiation of VSMCs
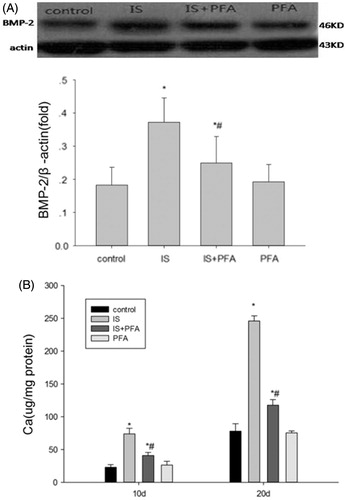
Because IS-promoted Pit-1 protein and mRNA expression, and Pit-1 itself is critical for calcification of VSMCs, we hypothesized that this function may participate in IS-induced osteoblastic differentiation of VSMCs. Thus, we examined the role of Pit-1 in IS-induced osteoblastic differentiation of VSMCs. The VSMCs were treated with 500 μM IS and 0.5 mM PFA, a competitive inhibitor of the NA-dependent Pi cotransporter. Expression of BMP-2, an osteoblastic differentiation marker, was examined. Western blot analysis demonstrated that PFA attenuated the IS-induced osteoblastic differentiation of VSMCs (). PFA itself did not induce osteoblastic differentiation as reported previously.Citation13 Furthermore, VSMCs were treated with 500 μM IS in the presence or absence of 0.5 mM PFA for 10 and 20 days to examine the calcium content, and with 500 μM IS and 0.5 mM PFA for 14 days to examine calcification. We found decreases in the calcium content and calcification of cells by treatment with PFA (). This result indicated that PFA could partly abolish IS-induced calcium deposition.
Figure 3. The role of Pit-1 in IS-induced osteoblast differentiation of VSMCs. (A) Top panels: representative western blots of BMP-2 when incubated VSMCs with 500 μM IS and 0.5 mM PFA. Graphs: quantification of the expression. (B) Calcium content of VSMCs cocultured with PFA, for 10 or 20 days with the indicated treatment. Results are presented as percentage of control values and are mean ± SD of three independent experiments. *p < 0.05, compared with the control group. #p < 0.05, compared with the IS-stimulated group.
The role of the JNK pathway in stimulation of Pit-1 expression by IS
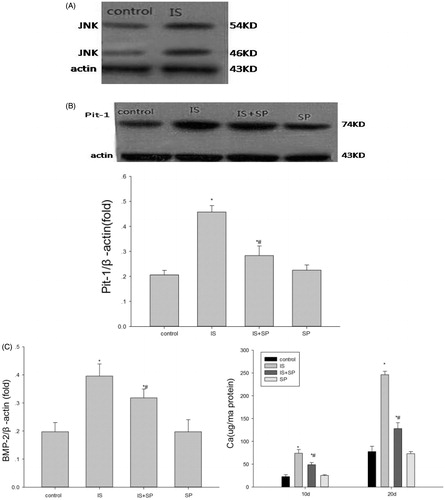
It has been reported that IS activates the JNK pathway in proximal tubularcells.Citation24 Additionally, Suzuki et al. demonstrated that, in BMP-2-induced calcification of osteoblast-like cells, a specific inhibitor of the JNK pathway decreases Pit-1 expression induced by BMP-2 without influencing basal Pit-1 activity.Citation25 Thus, we examined the expression of JNK after treatment of VSMCs with 500 μM IS. Compared with the control group, the expression of JNK was obviously enhanced (). Next, VSMCs were treated with 500 μM IS and the JNK inhibitor SP (20 μM). SP suppressed the IS-induced up-regulation of Pit-1 expression (). Furthermore, SP decreased calcium deposition and BMP-2 expression compared with cells treated with 500 μM IS only ().
Figure 4. The role of the JNK pathway in the upregulation of Pit-1 expression induced by IS. (A) The activation of JNK by 500 μM IS. (B) The JNK inhibitor, SP600125 (SP; 20 μM), partly suppressed the effect of IS on stimulation of Pit-1 expression. (C) SP partially suppressed the effect of IS on stimulation of BMP-2 expression and calcium content. Results are presented as percentage of control values and are mean ± SD of three independent experiments. *p < 0.05, compared with the control group. #p < 0.05, compared with the IS-stimulated group.
Discussion
We investigated the mechanisms by which IS enhances matrix mineralization in cultured VSMCs. IS treatment promoted osteoblastic differentiation, as previously reported,Citation23 as well as matrix mineralization. To clarify the mechanism, based on the crucial role of Pit-1 in calcification, we examined the expression of Pit-1 after IS treatment and observed upregulation. Next, we investigated the role of Pit-1 in IS-induced osteoblastic differentiation of VSMCs. Inhibition of Pit-1 by PFA attenuated the IS-stimulated osteoblastic differentiation and calcium deposition, indicating that upregulation of Pit-1 may be involved in IS-stimulated osteoblastic differentiation and calcium deposition. Furthermore, we inhibited the JNK pathway, which was activated by IS, and found suppression of the Pit-1 upregulation. Additionally, the IS-stimulated osteoblastic differentiation and calcium deposition were suppressed. Our study is the first to demonstrate that IS-promoted Pit-1 expression in part by activation of the JNK pathway, which is involved in the mechanism of IS-induced osteoblast differentiation and matrix mineralization.
IS, a common protein-bound uremic toxin, gradually accumulates in the body by renal functions. In addition to its nephrotoxicity, in recent years, IS has been found to have cardiovascular toxicity. IS is an independent risk factor for CVD in patients with CKD. A cross-sectional study of patients with stage 3–4 CKD showed that both inflammation and oxidative stress participate in IS-mediated cardiotoxicity,Citation26 which is in agreement with a study of hypertensive rats, showing that IS promotes cardiac fibrosis, which enhances oxidative stress.Citation22 It has also been demonstrated that IS stimulates arteriosclerosis by enhancing the expression of glucose transporter-1Citation27 and angiotensin II signallingCitation28 in VSMCs. In terms of calcification, Ayinuer et al. demonstrated that IS promotes aortic calcification and accelerates differentiation of VSMCs into osteoblast-like cells with expression of osteoblast-specific proteins in hypertensive rats.Citation21 Gulinuer et al. found that IS stimulates reactive oxygen species generation by upregulating the NADPH oxidase Nox4 and induction of osteoblast-specific proteins, such as Cbfa1, alkaline phosphatase, and osteopontin, in VSMCs,Citation23 which is consistent with our results. Furthermore, we corroborated that IS not only stimulated the expression of osteoblast-specific proteins in a time- and dose-dependent manner, but also induced matrix mineralization in vitro. Moreover, in patients with stage 4–5 CKD, vascular calcification is significantly alleviated by treatment with an oral sorbent (AST-120),Citation29 which reduces serum and urine levels of IS.Citation30 This finding indicates that IS might play an important role in calcification in vivo. However, the mechanism of IS-enhanced osteoblastic differentiation is still poorly understood. In this study, we explored the mechanism by which IS leads to calcification and oxidative stress. It has been reported that Pi uptake via Pit-1 is required for VSMC calcification, suggesting a crucial role of Pit-1 in osteogenic differentiation and mineralization of VSMCs.Citation12 Studies have found that changes in the expression level of Pit-1 regulate vascular calcification.Citation13,Citation14 Moreover, it has been reported that Pit-1 is involved in apoptosis of HeLa cells in response to tumor necrosis factor-α (TNF-α),Citation31 and that IS induces apoptosis of HK-2 cells,Citation32 indicating that IS may influence Pit-1 expression. Hence, we examined the expression of Pit-1 by the treatment of VSMCs with IS and found upregulation of Pit-1 expression at both protein and mRNA levels. Additionally, when Pit-1 was inhibited by PFA, we observed attenuation of osteoblast-specific protein expression and calcium deposition. Because a specific inhibitor of the JNK pathway reduces Pit-1 expression induced by BMP-2 without influencing basal Pit-1 activity in osteoblast-like cellsCitation22, we investigated the role of the JNK pathway in the upregulation of Pit-1 expression induced by IS. Similarly, we demonstrated that IS-promoted Pit-1 expression, at least partially, through activation of the JNK pathway.
The mean serum level of IS has been reported to be 249 μMCitation33 and 360 μM,Citation34 and the maximum serum level of IS is 557 μM in hemodialysis patients. We found that the influence of IS on matrix mineralization and Pit-1 expression was only partially time and dose dependent. This observation may be because IS is a uremic toxin and may lead to cell death at increasing concentrations. In our study, the effect of IS on matrix mineralization may have reached a plateau at 500 μM in vitro. Because the maximum serum level of IS is 557 μM, it may reach its maximum effect on matrix mineralization in vivo. Nonetheless, more evidence is required to validate this notion. The occurrence of calcification in patients with CKD is complex because it is influenced by many factors including traditional risk factors, such as age, heredity, smoking, hypertension and diabetes mellitus, and untraditional risk factors, such as calcium and phosphorus metabolism disorders, a chronic inflammatory state, and uremic toxins. Although we have demonstrated that IS promotes calcification partly through enhancing Pit-1 expression in vitro, whether it is involved in calcification in vivo is still unknown. Hence, further experiments are required. Moreover, further research is needed to determine whether high phosphorus-induced calcification would be aggravated under IS stimulation. The fact that IS enhanced the expression of Pit-1 in VSMCs does not indicate that it would occur under the influence of multiple stimulating factors or in vivo. Finally, when we blocked the JNK pathway, the effect of IS on Pit-1 expression was only partially attenuated, suggesting that there may be another mechanism involved in this process. However, Salaun et al. reported that hyper activation of JNK is involved in the increased sensitivity of Pit-1-depleted cells to apoptosis in response to TNF.Citation31 It appears that JNK is downstream of Pit-1 in the apoptosis of HeLa cells, which is not consistent with our study. This may be because of the different models and stimulators in each study. Additionally, it has been reported that activation of NF-κB may upregulate Pit-1 expression,Citation31 and that IS activates NF-κB in HK-2 cells.Citation32 It is unclear whether IS upregulates Pit-1 expression through the NF-κB pathway in VSMCs. Furthermore, while the JNK pathway was blocked, both BMP-2 expression and calcium deposition were attenuated. Nevertheless, there is no evidence that can elucidate the role of the JNK pathway in IS-induced osteoblastic differentiation and matrix mineralization of VSMCs. It is unclear whether IS activated the JNK pathway immediately or whether IS activated the JNK pathway to promote Pit-1 expression for osteoblastic differentiation and matrix mineralization. Further study, such as overexpression of Pit-1, is needed to explore this issue.
Conclusions
We found that IS treatment resulted in osteoblastic differentiation and matrix mineralization of VSMCs. The effects of IS were partly mediated by upregulation of Pit-1 expression, which was partially induced by activation of the JNK pathway. This study elucidated a novel mechanism underlying the pro-calcifying action of IS on VSMCs.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119.
- Vanholder R, Massy Z, Argiles A, Spasovski G, Verbeke F, Lameire N. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol Dial Transplant. 2005;20:1048–1056.
- Dhingra R, Sullivan L, Fox C, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167:879–885.
- Kestenbaum BR, Adeney KL, de Boer IH, Ix JH, Shlipak MG, Siscovick DS. Incidence and progression of coronary calcification in chronic kidney disease: The multi-ethnic study of atherosclerosis. Kidney Int. 2009;76:991–998.
- Tonelli M, Curhan G, Pfeffer M, et al. Relation between alkaline phosphatase, serum phosphate, and all-cause or cardiovascular mortality. Circulation. 2009;120:1784–1792. DOI: 10.1161
- Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–E17.
- Lau WL, Pai A, Moe SM, Giachelli CM. Direct effects of phosphate on vascular cell function. Adv Chronic Kidney Dis. 2011;18:105–112.
- Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ Res. 2011;109:697–711.
- Gonzalez M, Martínez R, Amador C, Michea L. Regulation of the sodium-phosphate cotransporter Pit-1 and its role in vascular calcification. Curr Vasc Pharmacol. 2009;7:506–512.
- Boyer CJ, Baines AD, Beaulieu E, Beliveau R. Immunodetection of a type III sodium-dependent phosphate cotransporter in tissues and OK cells. Biochim Biophys Acta. 1998;1368:73–83.
- Kavanaugh MP, Miller DG, Zhang W, et al. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci USA. 1994;91:7071–7075.
- Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–912.
- Li X, Yang HY, Giachelli CM. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis. 2008;199:271–277.
- Zavaczki E, Jeney V, Agarwal A, et al. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011;80:731–739.
- Derici U, E Nahas AM. Vascular calcifications in uremia: Old concepts and new insights. Semin Dial. 2006;19:60–68.
- Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:1551–1558.
- Lin CJ, Liu HL, Pan CF, et al. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res. 2012;43:451–456.
- Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;29:551–557.
- Watanabe I, Tatebe J, Namba S, Koizumi M, Yamazaki J, Morita T. Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells. Circ J. 2013;77:224–230.
- Taki K, Tsuruta Y, Niwa T. Indoxyl sulfate and atherosclerotic risk factors in hemodialysis patients. Am J Nephrol. 2007;27:30–35.
- Adijiang A, Goto S, Uramoto S, Nishijima F, Niwa T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol Dial Transplant. 2008;23:1892–1901.
- Yisireyili M, Shimizu H, Saito S, Enomoto A, Nishijima F, Niwa T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013;92:1180–1185.
- Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051–2058.
- Shimizu H, Bolati D, Higashiyama Y, Nishijima F, Shimizu K, Niwa T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-κB, p53, ERK, and JNK in proximal tubular cells. Life Sci. 2012;90:525–530.
- Suzuki A, Ghayor C, Guicheux J, et al. Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21:674–683.
- Rossi M, Campbell KL, Johnson DW, et al. Protein-bound uremic toxins, inflammation and oxidative stress: Across-sectional study in stage 3-4 chronic kidney disease. Arch Med Res. 2014;45:309–317.
- Lin CY, Hsu S.C, Lee HS, et al. Enhanced expression of glucose transporter-1 in vascular smooth muscle cells via the Akt/tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR)/ribosomal S6 protein kinase (S6K) pathway in experimental renal failure. J Vasc Surg. 2013;57:475–485.
- Shimizu H, Hirose Y, Goto S, et al. Indoxyl sulfate enhances angiotensin II signaling through upregulation of epidermal growth factor receptor expression in vascular smooth muscle cells. Life Sci. 2012;91:172–177.
- Goto S, Kitamura K, Kono K, Nakai K, Fujii H, Nishi S. Association between AST-120 and abdominal aortic calcification in predialysis patients with chronic kidney disease. Clin Exp Nephrol. 2013;17:365–371.
- Niwa T, Nomura T, Sugiyama S, Miyazaki T, Tsukushi S, Tsutsui S. The protein metabolite hypothesis, a model for the progression of renal failure: An oral adsorbent lowers indoxyl sulfate levels in undialyzed uraemic patients. Kidney Int. 1997;52:S23–S28.
- Salaun C, Leroy C, Rousseau A, Boitez V, Beck L, Friedlander G. Identification of a novel transport-independent function of PiT1/SLC20A1 in the regulation of TNF-induced apoptosis. J Biol Chem. 2010;285:34408–34418.
- Bolati D, Shimizu H, Yisireyili M, Nishijima F, Niwa T. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-κB. BMC Nephrol. 2013;14:56.
- Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med. 1994;124:96–104.
- Niwa T, Miyazaki T, Tsukushi S, et al. Accumulation of indoxyl-beta-D-glucuronide in uremic serum: Suppression of its production by oral sorbent and efficient removal by hemodialysis. Nephron. 1996;74:72–78.