
Abstract
This study aimed to explore the influence of neutrophil gelatinase-associated lipocalin on autophagy and its role in ischemia/reperfusion injury in human kidney-2 (HK-2) cells during acute kidney injury (AKI). HK-2 cells were given hypoxia/reoxygenation treatment for different times to simulate ischemia/reperfusion injury. Autophagy was evaluated by western blot and immunofluorescence of GFP-LC3. Cell viability was tested to reflect the degree of cell damage. The autophagy inhibitor 3-MA was used to inhibit autophagy and determine the role of autophagy in ischemia/reperfusion injury. HK-2 cells were hypoxia for 1 h, followed by reoxygenation treatment for 24 h. These cells were then exposed to human recombinant protein neutrophil gelatinase-associated lipocalin (NGAL) (50, 100, 200, 400, or 1000 ng/mL) with or without 3-MA. Our results showed that autophagy was induced by hypoxia treatment and was further enhanced by reoxygenation after hypoxia treatment. Cell viability was decreased with the inhibition of autophagy in the process. Autophagic flux was further induced with NGAL (>200 ng/mL), while cell viability declined in this condition. Cell viability was recovered when autophagy was inhibited. These results indicate that autophagy plays, in part, a protective role in renal ischemia/reperfusion injury. Furthermore, the data suggest that NGAL strengthens the level of autophagy in this process. Overall, a large quantity of NGAL produced by renal proximal tubular epithelial cells may induce excessive autophagy and increase renal ischemia/reperfusion injury in acute kidney injury.
Introduction
Acute kidney injury (AKI) is a severe kidney disease characterized by a rapid loss of renal function, which can result in an accumulation of metabolic wastes and an imbalance of electrolytes and body fluid. Renal ischemia/reperfusion (I/R) injury is a common risk factor for AKI. Recently, many studies have shown that autophagy is upregulated and plays an important role in I/R-induced AKI. Autophagy has been shown to protect renal tubular epithelial cells against I/R injury by clearing aging or damaged cellsCitation1–3 and also demonstrating a renoprotective role in AKI.Citation4–7 Conversely, several studies have reported on a supporting role of autophagy in tubular cell death in AKI.Citation8–10 Whether autophagy protects renal proximal tubular epithelial cells in AKI remains controversial.Citation10,Citation11
Autophagy is an accumulative degradation process in which cytosolic proteins and organelles are sequestrated by double-membrane structures of unknown origin (termed autophagosomes).Citation12 The proteins and organelles are then transferred to lysosomes and degraded by proteases.Citation12 Autophagy is induced under stressful conditions – including cell starvation, growth factor deprivation, hypoxia, and oxidant injury – and plays an adaptive role that ensures cell survival.
Neutrophil gelatinase-associated lipocalin (NGAL) is one of the maximally early induced genes in the post-ischemic kidney. Previous studies have indicated that NGAL may represent a novel early urinary biomarker for ischemic renal injury.Citation13 Moreover, NGAL has been shown to act as a cell-protective factor against hypoxia/reoxygenation in AKI via the regulation of the B-cell lymphoma 2 (Bcl-2) gene, hence protein regulation of Bcl-2 and Bax family members and caspase-3.Citation14 We hypothesized that in AKI, NGAL protects or damages cells by regulating autophagy.
Materials and methods
Cell culture and I/R injury
The human renal proximal tubular epithelial cell line HK-2 was cultured as a monolayer in Dulbecco’s Modified Eagles Medium: Nutrient Mixture F-12, supplemented with 10% (v/v) heat-inactivated fetal bovine serum at 37 °C in a humidified atmosphere containing 5% CO2.
To mimic ischemic injury in vitro, HK-2 cells were incubated in glucose-free medium in an anaerobic bag under hypoxia conditions. To model reperfusion injury, HK-2 cells were treated with a high concentration of glucose (55 mmol/L) culture medium at 37 °C in a humidified atmosphere containing 5% CO2 after ischemic treatment.
Drug treatments
To determine the effect of NGAL on autophagy in renal I/R, different concentrations of human NGAL recombinant protein (50, 100, 200, 400, and 1000 ng/mL) were added to the medium during I/R. Chloroquine (CQ) inhibits autophagy by blocking the fusion of the lysosome and autophagosome. CQ (10 μmol/L) was used in the present study to monitor autophagic flux. To examine the effect of autophagy on I/R-induced cell death, the autophagy inhibitor 3-methyladenine (3-MA, 5 mM) was added to the medium 1 h before each experiment.
Western immunoblot
HK-2 cells were lysed for 30 min at 4 °C with 15 μL of Radio-Immunoprecipitation Assay (RIPA) buffer (1% NP40, 0.5% sodium deoxycholate, and 0.1% sodium dodecylsulfate [SDS] in phosphate-buffered sodium solution, pH 7.4) containing protease inhibitors and then repeated three times for 10 seconds. Protein was determined by the bicinchoninic acid (BCA) assay. Samples were boiled (5 min) in loading buffer containing 4% SDS, 20% glycerol, and bromophenol blue. Protein of HK-2 cells were resolved via 10% SDS-polyacrylamide gel electrophoresis and then transferred to polyvinyl difluoride membranes (200 mA, 2 h, 4 °C). The membranes were blocked with 5% (M/V) non-fat milk in Tris-buffered saline (TBS) solution (25 mmol/L of Tris-HCl [pH 7.5] and 150 of mmol/L NaCl) containing 0.05% Tween for 1 h at room temperature. The membranes were then incubated with anti-LC3 (1:1000) or anti-P62 (1:1000) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1000) primary antibodies overnight at 4 °C, followed by washing with TBS solution plus 0.05% Tween. Membranes were then incubated with fluorescein isothiocyanate-conjugated secondary antibodies (1:10,000) for 2 h at room temperature via ECL detection. The intensities and optical densities of scanned immunoreactive protein bands were quantified and corrected by background subtraction and normalization to the intensity of GAPDH bands.
GFP-LC3 plasmids were transfected into HK-2 cells to examine the number of autophagosome by fluorescent microscope.
Cell viability assay
Cell viability was evaluated by measuring the fluorescence of resorufin using the CellTiter-Blue® cell viability assay kit according to the instructions of the manufacturer.
Statistical analysis
Results are expressed as the mean ± SD and were analyzed using the one-way analysis of variance (ANOVA) followed by the Newman–Keuls multiple comparison test. Statistical significance was reached when p < 0.05. All data were processed using IBM SPSS Statistics software version 19.0 (SPSS Inc., Chicago, IL).
Results
Autophagy plays a protective role in HK-2 cells during I/R injury
Autophagy-related protein LC3-II was tested by western immunoblot 1, 3, 6, 9, and 12 h after ischemia. LC3-II was significantly elevated after ischemia 1 h; however, a declining trend was observed following this time point (). HK-2 cells were treated with reperfusion 2, 12, and 24 h after hypoxia. LC3-II levels increased with 24 h of reperfusion compared with ischemia alone (). GFP-LC3 was expressed in HK-2 cells with transient transfection of the GFP-LC3 plasmids before I/R treatment. Extensive punctuate staining of GFP-LC3 was observed after ischemia. Quantification revealed that the punctate staining increased after I/R treatment (). Overall, these data showed that autophagy can be induced by ischemia and is heightened by I/R.
Figure 1. Autophagy in HK-2 cells during in vitro I/R and the cell viability after 3-MA treatment. (A) LC3-II expression was determined by Western blotting for different ischemic time. (B) LC3-II levels for different reperfusion time. (CV) Quantification of LC3-II levels in (A). (D) Quantification of LC3-II levels in (B). (E) The GFP-LC3 puncta formation in HK-2 cells was determined by immune fluorescence in ischemia and I/R. (F) LC3-II expression after HK-2 cells were treated by 3-MA in ischemic step. (G) LC3-II expression after HK-2 cells were treated by 3-MA in I/R. (H) The cell viability in ischemic condition after treated by 3-MA. (I) The cell viability after HK-2 cells were treated by I/R and 3-MA. In (C), (D), (H), and (I), data are presented as the means ± SDs in three independent experiments. *p < 0.05 as compared with the control and I1/R0 group. In (H) and (I), **p < 0.05 as compared with the group without 3-MA treated.
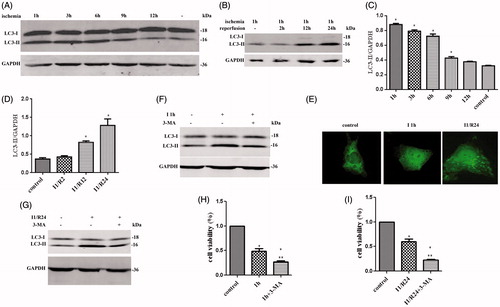
3-MA was then used to elucidate the role of autophagy in ischemia and I/R (). Treatment with 3-MA inhibited autophagy with a reduction in cell viability during ischemia and I/R (). These results indicated a protective role of autophagy in HK-2 cells during ischemia as well as in I/R.
NGAL exacerbates I/R injury in HK-2 cells by excessive autophagy
To investigate the effect of NGAL on autophagy in I/R, HK-2 cells were treated with recombinant protein NGAL/Lipocalin-2 and the expression of the autophagy marker LC3-II was examined by western immunoblot.
Low concentrations of NGAL (<200 ng/mL) did not enhance the level of LC3-II during I/R, while high concentrations (≥200 ng/mL) further increased the level of this protein ().
Figure 2. NGAL can further strengthened autophagy flux and aggravate cell injury. (A) LC3-II levels for different concentration NGAL. (B) Quantification of LC3-II levels in (A). (C) P62 and LC3-II levels after NGAL and CQ treated. (D) The GFP-LC3 puncta formation in HK-2 cells after I/R and NGAL treated. After CQ applied, the autophagosome accumulated to green lump. (E) The cell viability after NGAL and 3-MA treated. In (B) and (E), data are presented as the means ± SDs in three independent experiments. *p < 0.05 as compared with the control and I/R group. In (E), **p < 0.05 as compared with the group which without 3-MA treated.
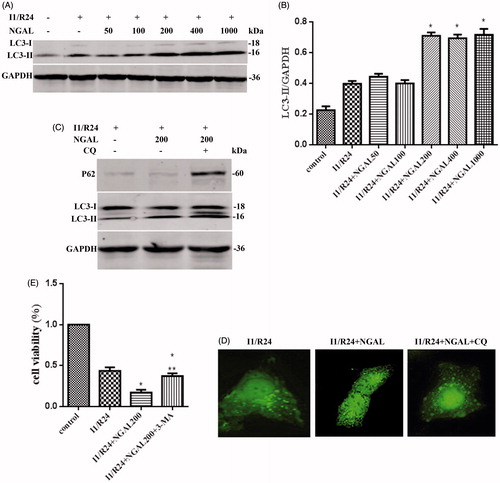
CQ was then added to test NGAL-induced autophagic flux through protein expression of p62 and LC3-II and GFP-LC3 staining (). Treatment of HK-2 cells exposed to NGAL and 3-MA increased cell viability compared with cells without 3-MA. These results suggested that NGAL-induced excessive autophagy is harmful to HK-2 cells during I/R ().
Discussion
Our data showed that autophagy in HK-2 cells was maximally induced with 1 h of ischemia and was not further heightened after this time point. Therefore, chronic ischemia in this study may have resulted in an over-consumption and degradation of the autophagosome. Furthermore, the autophagic regulatory molecules, such as ATG3, AMBRA1, and Beclin-1, may have been inhibited by caspase to compensate for the chronic stress state. The activity of mTOR may have also been reactivated during the chronic hypoxia.Citation15,Citation16 After 1 and 24 h of ischemic and reperfusion, respectively, autophagy was further enhanced. Although different signaling pathways for both stages have been previously shown to occur with enhanced autophagy,Citation17,Citation18 the underlying mechanism still requires further research. Therefore, the present study sought to study the role of autophagy in ischemia and reperfusion using 3-MA.
Our findings revealed that inhibition of autophagy with 3-MA was also accompanied by a sharp decline in cell viability during the ischemic stage of with I/R. This result confirmed that autophagy plays a renoprotective role in renal I/R, and corroborates previous findings observed in an animal model of I/RCitation19. In contrast, autophagy has been shown to protect myocardial cells in the ischemia phase, while it increases cell damage in the reperfusion phase.Citation20 Therefore, the role of autophagy may be dependent on different tissues and organs.
An important finding in this study was that autophagic flux was exacerbated by the elevated level of NGAL. The proteins p62 and LC3-II were shown to be involved because lysosomal degradation of LC3-II in the autophagosomes was blocked with CQ. This effect resulted in the inhibition of autophagic flux and an increase in the expression of LC3-II. If the increase of LC3-II by NGAL resulted from blocking the lysosomal degradation of LC3-II, the LC3-II band would, therefore, not be further enhanced by CQ. P62 is known to be degraded specifically by autophagic pathways and one of the key autophagic substrates. During an efficient flux of the autophagic pathway, p62 interacts with LC3 through its LC3-interacting domain. This enables the incorporation of p62 and p62-bound poly ubiquitinated protein aggregates into the autophagosome and thus, the p62–LC3 complex and bound aggregates are subsequently degraded by the lysosome. In the present study, CQ-mediated inhibition of autophagy flux resulted in the prevention of p62 and LC3 degradation by the autophagic-lysosomal pathway. This result may implicate these two proteins as markers to assess autophagic flux under certain conditions. We also found elevated levels of p62 and LC3-II in response to the accumulation of autophagosome after combined treatment of NGAL and CQ, thereby demonstrating that NGAL may induce an unbroken autophagy flux.
Our findings also showed that cell viability sharply decreased with NGAL treatment, and this effect was reversed with 3-MA. Under ischemic conditions, cells may undergo autophagy and apoptosis, and both events may be mutually induced and mutually restricted.Citation21 Excessive autophagy can result in various mechanisms of cell death, such as apoptosis, necrosis, and autophagy, which can act alone or together. Autophagy in the present study was induced during the ischemic stage and was further enhanced after I/R. We showed that a high concentration of NGAL significantly improved cell autophagy. It induces cell apoptosisCitation19 and type II programed cell death which is autophagic cell death.Citation20 Overall, our results suggest that NGAL treatment induces excessive autophagy and exacerbates cell damage resulting in cell death.
Funding information
The authors thank the support of the National Natural Science Foundation of China (Nos. 81271903 and 61201093).
Disclosure statement
The authors declare that we have no financial and/or personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled “Neutrophil gelatinase-associated lipocalin worsens ischemia/reperfusion damage of kidney cells by autophagy”.
References
- Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913.
- Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283.
- Liu S, Hartleben B, Kretz O, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia–reperfusion injury. Autophagy. 2012;8:826–837.
- Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia–reperfusion injury. Am J Pathol. 2010;176:1181–1192.
- Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640.
- Pallet N, Bouvier N, Legendre C, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4:783–791.
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–F787.
- Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int. 2009;76:604–613.
- Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation. 2007;84:1183–1190.
- Suzuki C, Isaka Y, Takabatake Y, et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun. 2008;368:100–106.
- Gotoh K, Lu Z, Morita M, et al. Participation of autophagy in the initiation of graft dysfunction after rat liver transplantation. Autophagy. 2009;5:351–360.
- Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175.
- Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–2543.
- Cui LY, Yang S, Zhang J. Protective effects of neutrophil gelatinase-associated lipocalin on hypoxia/reoxygenation injury of HK-2 cells. Transplant Proc. 2011;43:3622–3627.
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: The interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94.
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946.
- Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190.
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922.
- Rami A, Kogel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy. 2008;4:422–426
- Baehrecke EH. Autophagic programmed cell death in Drosophila. Cell Death Differ. 2003;10:940–945.
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975.