
Abstract
Ischemia is the most frequent cause of acute kidney injury (AKI), which is characterized by apoptosis of renal tubular cell. A common result of ischemia in AKI is dysfunction of endoplasmic reticulum (ER), which causes the protein-folding capacity to lag behind the protein-folding load. The abundance of misfolded proteins stressed the ER and results in induction of the unfolded protein response (UPR). While the UPR is an adaptive response, over time it can result in apoptosis when cells are unable to recover quickly. Recent research suggests that ER stress is a major factor in renal tubular cell apoptosis resulting from ischemic AKI. Thus, ER stress may be an important new progression factor in the pathology of ischemic AKI. In this article, we review UPR signaling, describe pathology and pathophysiology mechanisms of ischemic AKI, and highlight the dual function of ER stress on renal tubular cell apoptosis.
Introduction
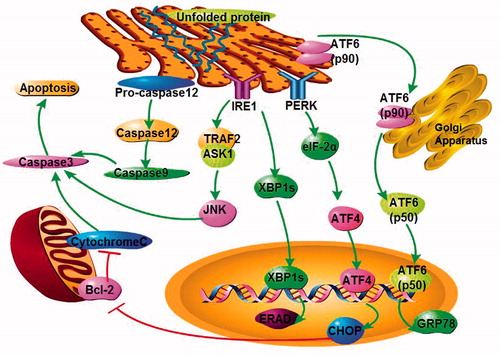
Endoplasmic reticulum (ER) stress can be triggered by many different stimulatory signals in ischemic acute kidney injury (AKI), including mutant protein aggregation, hypoxia, energy deprivation, and metabolic dysfunction. Declined protein-folding capacity in ER leads to abundance of misfolded proteins, initiating ER stress. Overwhelming ER stress causes apoptosis through the three typical signal pathways, PERK–eIF2–ATF4 pathway, IRE1–XBP1 pathway and ATF6 pathway (). The ability of renal tubular cells to cope with ER stress is essential for maintaining normal renal function. Therefore, a deeper understanding of the pathological contribution of ER stress and its interaction with key underlying mechanisms will advance our ability to fully recognize the disease.
Figure 1. ER stress pathways. The accumulation of misfolded proteins in the ER activates the three typical signal pathways, PERK–eIF2–ATF4 pathway, IRE1–XBP1 pathway and ATF6 pathway as well as caspase 12.
Endoplasmic reticulum stress
Secretory and membrane proteins are synthesized and folded in the ER, where these proteins also receive post-translational modifications such as glycosylation, disulfide bond formation and lipidation.Citation1 Before proteins exit the ER and are transported to the Golgi and other destinations within the cell, they must be correctly folded into a specific final structure.Citation2 Normal proteins achieve the correct structure and physiological function following the correct folding process. However, if proteins manufactured in the ER are prevented from attaining their proper tertiary structure, they become misfolded. These misfolded proteins can aggregate, and the accumulation of misfolded proteins in the ER will result in cellular stress and finally cellular damage, a process which is referred to as ER stress.Citation3 The immediate response to ER stress is the activation of intracellular signal transduction pathways in ER, collectively called the unfolded protein response (UPR).Citation4 ER stress can be triggered by various stimuli, such as ischemia, hypoxia, oxidative stress, glucose starvation, and elevated protein synthesis.Citation3,Citation5–7 Three major ER stress response transmembrane proteins are primarily activated, including protein kinase-like ER kinase (PERK), inositol requiring 1 (IRE1), and activating transcription factor 6 (ATF6), which activate downstream signaling effectors.
GRP78 is a negative regulator of the UPR
Glucose-regulated protein 78 (GRP78), also called Immunoglobulin binding protein (BiP), is a molecular chaperone in cells, which can promote protein folding with hydrolysis of ATP. GRP78 binds to the unfolded or incompletely folded proteins and prevents interactions of misfolded proteins with surrounding molecules within the ER lumen. In unstressed cells, GRP78 is bound to PERK/IRE1/ATF6 to keep them in an inactive state. When it encounters accumulated misfolded proteins in the ER, GRP78 disassociates from the three inducers of the UPR to help protein folding.Citation8 Consequently, released PERK/IRE1/ATF6 undergo activation, which leads to the initiation of the UPR.Citation9 In this way, GRP78 is a negative regulator of the UPR and plays a critical role in its initiation.
ATF6 signaling pathway
Upon dissociation from GRP78, Golgi localization signals on ATF6 (p90) are uncovered. ATF6 (p90) subsequently moves to the Golgi and is cleaved by Golgi-resident site-1 and site-2 proteases, releasing the cytosolic domain of ATF6 (p60). The cleaved activating transcription factor 6 (cATF6) contains a DNA-binding domain. Upon translocation to the nucleus, transcription of its target genes, including ER chaperones and enzymes such as XBP1 and CHOP/GADD153, is activated. Thus, ATF6 functions as a critical regulator of ER quality control proteins in mammalian cells.Citation10
IRE1 signaling pathway
IRE1 maintains is inactive when bound to GRP78 during homeostasis. Once ER stress occurs, GRP78 dissociates from IRE1 and triggers the activation of the endoribonuclease domain in IRE1, forming splicing X-box-binding protein-1 (sXBP-1) mRNA. sXBP-1 generates a functional XBP-1 protein that acts as a potent transcriptional activator to amplify the ER-associated degradation (ERAD) factors, the misfolded protein degradation system,Citation11 and the expression of ER resident chaperone GRP78. The IRE1–XBP1 pathway serves as an adaptive response to ER stress by degrading or refolding misfolded proteins accumulated in the ER lumen.Citation8
Activation of ATF6 precedes the activation of IRE1 during ER stress. In the initial stages of the UPR, ATF6 signaling pathway leads to accumulation of unspliced XBP1 mRNA. In parallel with ATF6, activated IRE1 is then available for splicing XBP1 mRNA. In addition, the cytosolic domain of IRE1 binds tumor necrosis factor receptor-associated factor 2 (Traf2), which then actives the c-Jun N-terminal Kinase (JNK)-mediated apoptotic pathway via apoptosis signal regulating kinase-1 (ASK1) phosphorylation.Citation12 This pathway, which is independent of XBP1, ultimately leads to apoptosis upon ER stress.
PERK signaling pathway
During ER stress, activation of PERK occurs by dimerization and trans-autophosphorylation, leading to the recruitment and phosphorylation of its substrate, eIF2a. Phosphorylation of eIF2a is inhibitory and reduces overall translation in cells, thereby decreasing the ER protein load. The attendant decrease in eIF2a activity paradoxically activates the translation of the ATF4 mRNA,Citation13 which can be preferentially translated. After ATF4 enters the nucleus, it activates the transcription of CHOP (C/EBP homologous protein), which can downregulate bcl-2 and cause cell apoptosis.Citation14 CHOP−/− mice and cells exhibited significantly less programed cell death,Citation15 and deregulated CHOP activity compromises cell viability.Citation14 On the other hand, ATF4 and CHOP can also activate GADD34 (growth arrest and DNA-damage inducible protein-34), promoting eIF2a dephosphorylation by the Protein Phosphatase 1 (PP1) complex thereby exerting negative feedback on the PERK pathway.Citation16
Prosurvival or proapoptotic effects of ER stress
The activation of the UPR induces an adaptive response in which the cell attempts to overcome the accumulation of misfolded proteins and ER stress. However, prolonged UPR activation has been demonstrated to have toxic effects, ultimately leading to cell death.Citation12 UPR involves factors that may lead to either cell survival or apoptosis, depending on the degree of ER stress. Prosurvival effects include eIF2a phosphorylation, GRP78 induction, and XBP splicing. Proapoptotic factors include JNK phosphorylation, CHOP/GADD153 induction, and caspase-12 activation. Caspase 12 is an ER-specific caspase, which helps initiate ER stress-mediated apoptosis through a caspase-9/caspase-3 cascade reaction upon activation.Citation17
Ischemic acute kidney injury
Acute kidney injury is characterized by a rapid decline of renal function, leading to the accumulation of metabolic waste and toxins and, even worse, resulting in failure of other organs. According to the Kidney Disease Improving Global Outcomes (KDIGO) working Group, AKI is defined as any of the following three criteria: (1) increase in serum creatinine (sCr) by ≥ 0.3 mg/dL (≥26.5 μmol/L) within 48 h or (2) an increase in serum creatinine to ≥1.5 times baseline, which is known or presumed to have occurred within the preceding 7 days; or 3) a urine volume <0.5 mL/kg/h for 6 h.Citation18,Citation19 AKI is a common problem among hospitalized patients, affecting around one in five patients admitted to hospital, with mortality rates ∼25–40% in severe cases.Citation20,Citation21 This is of critical importance to the elderly population, as the number of AKI patients’ increases with age.
AKI is a multifactorial renal disease, encompassing a wide spectrum of injury to the kidneys such as ischemia-reperfusion (IR) injury, sepsis, drugs, obstruction and various endogenous or exogenous injuries. AKI can be divided into pre-renal, intrinsic, and post-renal (obstructive) AKI. Intrinsic AKI can be further divided into tubulo-interstitial, glomerular, and vascular lesion.Citation22 Most tubulo-interstitial disease, and indeed most AKI, is caused by ischemia, which causes a generalized or localized impairment of both oxygen and nutrient delivery to cells and waste product removal from cells in the kidney.Citation19 In animal models, mice undergo unilateral clamping of the renal pedicle for a certain time and simultaneous contralateral nephrectomy to develop ischemic AKI.Citation23 In humans, renal surgery or transplantation, cardiac surgery, blood loss, blockages in kidney blood vessels, hypoperfusion, hypotension, and various other factors can all initiate ischemic AKI.Citation24–26
Changes of tubular epithelial cells in ischemic AKI
Pathologically, AKI is characterized by sublethal and lethal damage of renal tubular epithelial cells, endothelial cell injury, inflammation, and hemodynamic dysfunction.Citation27,Citation28 Injury and death of tubular cells are especially recognized as the characteristic pathological change in AKI. Furthermore, tubular repair and regeneration are considered as major events in kidney recovery from AKI. In ischemic AKI, there is an imbalance of local tissue oxygen supply and demand as well as accumulation of waste products of metabolism. As a consequence of this mismatch, the tubular epithelial cells undergo injury and with increasing time or severity of ischemia, there is cell death by either necrosis or apoptosis, resulting in impairment of water and electrolyte homeostasis and reduced excretion of waste products of metabolism.Citation29 Although sublethal injury is reversible, the death of tubular cells is accompanied by the inevitable loss of the function of the affected cells. Epithelial cell injury associated with ischemia is most apparent in the S3 segment of the proximal tubule.Citation30 Characteristic histological changes of renal tubular cells include effacement and loss of the tubular brush border, sloughing of tubular epithelial cells or the necrotic cell debris into the tubular lumen, the dilatation of the tubular lumen, and the formation of tubular casts due to necrosis and apoptosis.Citation29,Citation31,Citation32 Rather than necrosis, apoptosis may be the dominant mode of injury.Citation24 In the last two decades, tubular apoptosis has been demonstrated in various animal models and some clinical samples from patients with AKI. Experimentally, apoptosis can be observed by cell morphology, caspase activation, and terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) assay of DNA damage as well as the activation of other pro-apoptotic proteins, for example, CHOP and Bcl-2 family.
ER stress interacts with the extrinsic and intrinsic apoptosis signaling pathways
In tubular epithelial cells, stimulation of ischemia triggers the accumulation of unfolded proteins in the ER lumen, leading to the UPR. Early adaptive responses of UPR involve expansion of ER membranes, accelerated degradation of unfolded proteins, increased translation of folding chaperones, and inhibition of general protein synthesis.Citation33 However, if the damage is severe irreversible damage will occur and cells will inevitably undergo apoptosis.
Mechanistically, there are three main ways for inducing cell apoptosis: extrinsic pathways, intrinsic pathways, and ER stress. The intrinsic pathway is derived from nuclear DNA damage, ischemia, oxidative stress and so on, which leads to the oligomerization of Bax and Bak. This results in mitochondrial outer membrane permeability (MOMP), and the resulting release of Cytochrome C (Cyt C) causes activation of caspase 9 and caspase 3.Citation12,Citation34 The extrinsic pathway of apoptosis is initiated by death ligand binding to the death receptor (e.g. FasL binding to Fas), leading to formation of the Death inducing signaling complex (DISC) and activation of caspase 8. Activated caspase 8 amplifies the apoptotic cascade through cleaving Bid to its truncated form tBid, which can increase the release of the apoptogenic factor Cyt C from mitochondria,Citation35–37 eventually resulting in apoptosis.
In the ER stress pathway, JNK phosphorylation, CHOP/GADD153 induction, and caspase-12 activation can all lead to apoptosis. Activation of caspase-12 or JNK can activate the caspase-9/caspase-3 or caspase-3 cascades, respectively, which are important members in the intrinsic death pathway. CHOP promotes hyperoxidation of the ER and promotes IP3-induced Ca2 + release from the ER lumen.Citation38 Ca2 + leaked from the ER lumen enters the mitochondria and generates mitochondrial reactive oxygen species (ROS), triggering a vicious cycle of oxidative stress both in the ER and mitochondria.Citation39 In addition, CHOP decreases the expression of pro-survival protein BCL-2.Citation14 BCL-2 can inhibit the release of Cyt C from mitochondria, and therefore CHOP indirectly abolishes this protective effect.
Double effects of ER stress on renal tubular cells apoptosis in ischemic AKI
The early protective effects of ER stress
The increased amount of GRP78 protein after ischemia-reperfusion was mainly localized in the proximal tubule cells.Citation40 GRP78 elevates earlier than blood urea nitrogen(BUN) and creatinine, which reached their peaks 24–48 h after ischemia. The UPR was activated soon after renal ischemia, and this activation may have had a protective effect. For example, pretreatment with tunicamycin or thapsigargin, two ER stress inducers, protected tubule cells from renal ischemia-reperfusion injury through enhancement of GRP78 protein expression, and ameliorated renal dysfunction and injury.Citation40 In a culture system of simulated ischemia, proteomic analysis of renal tubule epithelial LLC-PK1 cells showed upregulation of GRP78 and heat shock protein 70(HSP70), which confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death.
Moreover, ischemic preconditioning (PC), defined as brief intermittent cycles of ischemia alternating with reperfusion applied after the ischemic event, may increase the tolerance of the ischemic kidneys against sustained injury.Citation41,Citation42 The use of ischemic PC protects kidney from ischemia-induced injury by suppressing ER stress, as indicated by downregulation of GRP78, ATF4, PERK, XBP-1 and the caspase12 protein levels.Citation43 Additional research reported that early ischemic PC attenuated ER stress in ischemic kidneys via increasing the relative amounts of GRP78 and decreasing PERK, ATF4 and TNF-receptor associated factor 2 (TRAF2) levels. The beneficial impact of early ischemic PC was dependent on nitric oxide (NO) levels, as the protective effect was abolished when NO production was inhibited before early ischemic PC application.Citation44 The early protective effects on ER stress could possibly be because ischemic PC leads to a pro-survival phenotype in different cells. In renal epithelial cells suffering from oxidative injury, preconditioning with ER stress caused activation of ERK1/2 signaling, accompanied by a reduction in JNK activation. Thus, the ER stress response modulates the balance between ERK and JNK signaling pathways to prevent cell death after oxidative injury.Citation45 Consistent with this, in the post-ischemic kidney, cells were protected from the following ischemia injury for up to 15 days and this was associated with reduction of JNK signaling by remote ischemic pretreatment.Citation46 Also of note, benefits related to ischemic PC have been documented in laboratory studies. However, the relevance to human AKI remains to be determined in a large number of clinical trials.
Apoptosis promoting effect of ER stress and interventions
Upregulation of mRNAs encoding the ER localized proteins GRP78, GRP94, and ERP72 were observed 30 min following acute renal ischemia.Citation47 In ischemia induced AKI mouse models, renal tubular necrosis score and cell apoptosis index reached their peak 24 hr after IR. GRP78, CHOP expression, and Caspase 12 activation were enhanced, reaching their peaks at 4 and 12 h, respectively.Citation48 A 20-fold increase in phospho-eIF2a accompanied by activation of the PERK pathway was observed in kidney homogenates following 10 min of cardiac arrest-induced ischemia and 10 min reperfusion.Citation25
In parallel, two ER stress hallmarks, GRP78 and CHOP, showed increased protein levels after hypoxia and ER stress, and participated in hypoxia/reoxygenation induced apoptosis in human renal proximal tubular epithelial cell line (HK-2 cells).Citation49 However, those parameters were significantly suppressed by berberine pretreatment.Citation49 It was also demonstrated that tauroursodeoxycholic acid (TUDCA) pretreatment had a nephroprotective effect on ischemia-induced AKI by inhibiting ER stress and by blocking GRP78 and CHOP expression, reducing Caspase 12 activation, and inhibiting cell apoptosis.Citation48
There are other interventions being explored. 1-(3,4-dihydroxyphenyl)-2-thiocyanate-ethanone, referred to as BiP inducer X (BIX), selectively upregulates GRP78/BiP mRNA and protein in cultured cells via the ATF6 pathway,Citation50 without inducing the XBP-1 and CHOP mRNAs, to protect against ischemic AKI.Citation51 Bax inhibitor-1 also has been shown to have a cytoprotective effect against renal ischemic injury by modulating ER stress. After ischemia, marked increases in sXBP-1, CHOP, ATF6, and phospho-JNK were found in kidneys in Bax inhibitor-1 knockout mice in comparison to wild type animals, indicating that injury and renal dysfunction was greater.Citation52 Moreover, the 150 kDa oxygen-regulated protein (ORP150) is an inducible ER chaperone protein with cytoprotective properties related to cell stresses such as ischemia. Renal tubular epithelial cells transfected with ORP150 were protected against hypoxic injury, and transgenic mice overexpressing ORP150 subjected to renal I/R displayed a blunted kidney injury.Citation53
Conclusion
The ER has many important roles in the induction of renal tubular epithelial cell apoptosis in ischemic AKI. Double effects of ER stress suggest that modulation of ER stress through pharmacologic intervention to amplify the cytoprotective aspects of the UPR, such as GRP78 and sXBP-1 upregulation, while down regulating the apoptosis promoting effects of UPR induction, such as CHOP and JNK upregulation, may benefit ischemic-related renal injury. Further explorations of the underlying mechanisms between ER stress and ischemic AKI are still needed.
Actually, the intrinsic, extrinsic and ER stress associated apoptotic pathways have all been implicated in ischemic AKI and have a lot of interrelation and interaction. For example, mitochondria is one of the converging points in cell apoptosis. In this regard, combination therapies that block multiple upstream pathways of cell apoptosis simultaneously or at different time points to ensure cell survival and renal function may be one of the future research focuses.
Disclosure statement
The authors declare that they have no competing interests. M.G. designed and drafted manuscript; J.Z. edited and revised manuscript; Y.X., W.J., H.D., Y.H. and X.-F. An approved final version of manuscript. This article does not contain any studies with human participants or animals performed by any of the authors.
Funding information
This study is funded by Natural Science Foundation of China (No. 81170688 and 81470973) and Shandong Province Natural Science Foundation (ZR2014CM040).
References
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63.
- Herrmann JM, Malkus P, Schekman R. Out of the ER-outfitters, escorts and guides. Trends Cell Biol. 1999;9:5–7.
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658.
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529.
- Badiola N, Penas C, Miñano-Molina A, et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149–e156.
- Inagi R, Nangaku M, Onogi H, et al. Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int. 2005;68:2639–2650.
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293.
- Inagi R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr Opin Pharmacol. 2010;10:156–165.
- Shen X, Zhang K, Kaufman RJ. The unfolded protein response-a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004;28:79–92.
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89.
- Vembar SS, Brodsky JL. One step at a time: Endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957.
- Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol. 2009;112:e1–e9.
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365.
- McCullough KD, Martindale JL, Klotz L-O, Aw T-Y, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259.
- Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995.
- Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077.
- Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985–1992.
- Bienholz A, Feldkamp T, Kribben A. Acute kidney injury. Dtsch Med Wochenschr. 2013;138:1229–1232.
- El Sabbahy M, Vaidya VS. Ischemic kidney injury and mechanisms of tissue repair. Wiley interdisciplinary reviews. Syst Biol Med. 2011;3:606–618.
- National Clinical Guideline C. National Institute for Health and Clinical Excellence: Guidance. In: Acute Kidney Injury: Prevention, Detection and Management Up to the Point of Renal Replacement Therapy. London: Royal College of Physicians (UK) National Clinical Guideline Centre; 2013.
- Uchino S, Bellomo R, Goldsmith D, Bates S, Ronco C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit Care Med. 2006;34:1913–1917.
- Kanagasundaram NS. Pathophysiology of ischaemic acute kidney injury. Ann Clin Biochem. 2015;52:193–205.
- Skrypnyk NI, Harris RC, de Caestecker MP. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Visual Exp. 2013;78:e50495–e50500.
- Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40.
- Montie HL, Kayali F, Haezebrouck AJ, Rossi NF, Degracia DJ. Renal ischemia and reperfusion activates the eIF 2 alpha kinase PERK. Biochim Biophys Acta. 2005;1741:314–324.
- Serviddio G, Romano AD, Gesualdo L, et al. Postconditioning is an effective strategy to reduce renal ischemia/reperfusion injury. Nephrol Dial Transplant. 2008;23:1504–1512.
- Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–2210.
- Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520.
- Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Investing. 2011;121:4210–4221.
- Lieberthal W, Nigam SK. Acute renal failure. I. Relative importance of proximal vs. distal tubular injury. Am J Physiol. 1998;275:F623–F631.
- Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–430.
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: Definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14.
- Herrmann AG, Deighton RF, Le Bihan T, et al. Adaptive changes in the neuronal proteome: Mitochondrial energy production, endoplasmic reticulum stress, and ribosomal dysfunction in the cellular response to metabolic stress. J Cereb Blood Flow Metab. 2013;33:673–683.
- Riedl SJ, Salvesen GS. The apoptosome: Signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413.
- Padanilam BJ. Cell death induced by acute renal injury: A perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol. 2003;284:F608–F627.
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–137.
- Sanz AB, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol. 2008;19:1634–1642.
- Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta. 2014;1843:2150–2163.
- Dejeans N, Tajeddine N, Beck R, et al. Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem Pharmacol. 2010;79:1221–1230.
- Prachasilchai W, Sonoda H, Yokota-Ikeda N, et al. A protective role of unfolded protein response in mouse ischemic acute kidney injury. Eur J Pharmacol. 2008;592:138–145.
- Chen H, Xing B, Liu X, et al. Similarities between ozone oxidative preconditioning and ischemic preconditioning in renal ischemia/reperfusion injury. Arch Med Res. 2008;39:169–178.
- Liu L, Lin YQ, Yan LT, et al. Extracellular ascorbic acid fluctuation during the protective process of ischemic preconditioning in rabbit renal ischemia-reperfusion model measured. Chin Med J. 2010;123:1441–1446.
- Mahfoudh-Boussaid A, Zaouali MA, Hauet T, et al. Attenuation of endoplasmic reticulum stress and mitochondrial injury in kidney with ischemic postconditioning application and trimetazidine treatment. J Biomed Sci. 2012;19:71–84.
- Mahfoudh-Boussaid A, Zaouali MA, Hadj-Ayed K, et al. Ischemic preconditioning reduces endoplasmic reticulum stress and upregulates hypoxia inducible factor-1alpha in ischemic kidney: The role of nitric oxide. J Biomed Sci. 2012;19:7–14.
- Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem. 2003;278:29317–29326.
- Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem. 2001;276:11870–11876.
- Kuznetsov G, Bush KT, Zhang PL, Nigam SK. Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc Natl Acad Sci USA. 1996;93:8584–8589.
- Gao X, Fu L, Xiao M, et al. The nephroprotective effect of tauroursodeoxycholic acid on ischemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin Pharmacol Toxicol. 2012;111:14–23.
- Yu W, Sheng M, Xu R, et al. Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med. 2013;11:24–33.
- Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15:364–375.
- Prachasilchai W, Sonoda H, Yokota-Ikeda N, et al. The protective effect of a newly developed molecular chaperone-inducer against mouse ischemic acute kidney injury. J Pharmacol Sci. 2009;109:311–314.
- Bailly-Maitre B, Fondevila C, Kaldas F, et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2006;103:2809–2814.
- Bando Y, Tsukamoto Y, Katayama T, et al. ORP150/HSP12A protects renal tubular epithelium from ischemia-induced cell death. FASEB J. 2004;18: 1401–1403.