
Abstract
Background: Focal segmental glomerulosclerosis (FSGS) is a frequent and severe glomerular disease characterized by destabilization of podocyte foot processes. Emerging evidence suggests that microRNAs play crucial roles in podocyte homeostasis. The aim of the present study was to determine the role of miR-206 in podocyte injury and to elucidate the mechanisms responsible for the damage to podocyte. Methods: FSGS nephropathy model was induced by a single intravenous injection of Adriamycin (ADR) in BALB/c mice and the levels of proteinuria were measured on week of 1,3,5. The conditionally immortalized mouse podocyte cell line 5 was either transfected with microRNA mimics or negative control. Real-time PCR analysis was conducted to demonstrate the microRNA level in the glomeruli. Expression of Wilms’ tumor-1 (WT1) and synaptopodin were detected by immunofluorescence and western blotting. Results: The results revealed that miR-206 was up-regulated in ADR nephropathy mice, which led to severe podocyte injury and inhibited the expression of WT1 and synaptopodin. Using luciferase reporter assays, WT1 was identified as a target gene of miR-206. In vitro, over expression of miR-206 induced WT1 and synaptopodin degradation and actin rearrangement, initiating a catastrophic collapse of the entire podocyte-stabilizing system. Conclusions: Over expression of miR-206 promotes podocyte injury via downregulation of WT1, which provides a new pathogenic mechanism for FSGS and miR-206 may be a potential therapeutic target.
Introduction
Focal segmental glomerulosclerosis (FSGS) is a disease of children and adults with unknown origin and clinically characterized by nephrotic-range proteinuria, frequent cause of steroid-resistant, leading to a further renal damage and increased cardiovascular morbidity and mortality.Citation1 It is diagnosed by renal biopsy, in which characteristic features are complete flattening of podocyte foot processes and foci of glomerular sclerosis. Recent studies have revealed that FSGS is the result of mutations in crucial podocyte genes.Citation2 Wilms’ tumor-1 (WT1) is a gene that is essential for the development and maintenance of normal podocyte and glomeruli.Citation3,Citation4 Loss of WT1 in pathologic conditions is commonly interpreted as a result of podocyte depletion.Citation5 However, exactly how WT1 is regulated in glomerular podocyte is essentially unknown. MicroRNAs (miRNAs) are a class of short and non-coding RNAs which play indispensible roles in the regulation of gene expression through suppressing their target genes by binding to complementary sequences and leading to inhibition of translation and mRNA degradation.Citation6,Citation7 Recent studies have indicated that miRNAs play a key role in kidney diseases.
In this context, we aimed to determine the roles and mechanisms of miR-206 in FSGS, which would be paramount for understanding the pathomechanisms of podocyte injury as well as for designing therapeutic strategies to combat proteinuric kidney disease.
Methods
Animal models
Male BALB/c mice, weighing approximately 20–22 g, were purchased from the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, China). For the Adriamycin (ADR) nephropathy model, BALB/c mice (n = 6) were administered by a single intravenous injection of ADR (SigmaAldrich, St. Louis, MO) at 10.5 mg/kg body wt.Citation8,Citation9 The mice in the control group received an equivalent volume of normal saline (NS). On week of 1, 3 and 5 following the administration of ADR or NS, individual mice were placed in metabolic cages for 24-h urine specimens’ collection. Urine specimens were tested for urinary albumin. After urine collection, groups of mice were euthanized and kidney tissues were collected for various analysis. The glomeruli were isolated using a magnetic bead perfusion method.Citation10 The study was approved by the Ethics Committee of Sun Yat-sen University Cancer Center.
Cell culture and treatment
The conditionally immortalized mouse podocyte cell line 5 (MPC5) was cultured as previously described.Citation11,Citation12 MPC5 cells were cultured at 33 °C (10 U/mL of interferon-γ) with 5% CO2 and were differentiated at 37 °C (without interferon-γ) with 5% CO2 in RPMI 1640 Dutch-modified medium (Invitrogen, Carlsbad, CA) supplemented with 10% v/v fetal calf serum. Depending on the exact experimental setup, the cultured cells were transfected with miRNA mimics or negative control using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY). In untreated groups (NT), cells were treated without any agents.
Dual-luciferase activity assay
The 3'UTR of the mouse WT1 cDNA, which contains a putative target site for miR-206, was amplified by PCR and inserted into the pGL3 control vector (Promega, Madison, WI) immediately downstream of the stop codon of the firefly luciferase gene (pGL3-WT1–3'UTR). A mutated version of the 3'UTR that contained a 7-bp mutation (3'mutant) was also generated using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). For the reporter assays, the pRL-SV40 vector (Promega) encoding Renilla luciferase was transiently cotransfected with a wild-type or mutant reporter plasmid containing the 3'UTR of WT1 and miRNAs using Lipofectamine 2000. The activities of both the firefly (Fluc) and Renilla (Rluc) luciferases were measured consecutively with a dual-luciferase assay kit (Promega, Madison, WI) at 24–48 h post-transfection.
RNA extraction and Real-time PCR
RNA was extracted using TRIzol reagent (Ambion, Austin, TX). Expression of miRNAs was detected by the TaqMan microRNA assay (Applied Biosystems, Foster City, CA), according to manufacturer’s instructions. The input was normalized by U6 snRNA. miR-206 and U6 primer sets were purchased from Applied Biosystems. For the detection of target mRNA, total RNA was reverse transcripted to cDNA by RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD) and subsequently amplified by PCR. Three independent experiments were performed, and the results were given as means ± SD.
Western Blot analysis of WT1 and synaptopodin
For Western Blot analysis, the specimens were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. Protein expression was analyzed with primary antibody against WT1 (1:1,000; Cell Signaling Technology, Beverly, MA), synaptopodin (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) together with an antibody against β-actin (1:10,000; Jackson Immuno Research, Philadelphia, PA) and then incubated with an appropriate secondary antibody. After washing, the protein was visualized with Super Signal Western Pico chemiluminescent substrate (Pierce, Rockford, IL), and signals were detected by an Odyssey Detection System (Li-COR, Lincoln, NE). Each of the above experiments was performed in triplicate.
Electron microscopy
For transmission electron microscopy, cortical tissues of kidney were dissected into 1–2 mm sections, double-fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated and embedded into Spurr’s epoxy resin (in Epon by standard procedures). Ultrathin sections (90 nm) were made and double-stained with uranyl acetate and lead citrate, and examined using a transmission electron microscope (JEM 100 CX-II, JEOL, Tokyo, Japan). Podocyte lesions including foot process effacement and basement membrane thickening were analyzed.
Histomorphometry, immunofluorescence and F-actin cytoskeleton staining
Histologically, paraffin-embedded sections (3-mm thickness) from the mouse kidney were stained with Masson’s trichrome and PAS by a routine procedure.
Immunohistologically, paraffin-embedded sections (6-mm thickness) from the mouse kidney were cut and dehydrated in xylene, followed by antigen retrieval in R-buffer A (EM Science, Hatfield, PA) by autoclave (ProteoGenix, Obershausbergen, France). Sections were then blocked with PBS containing 3% bovine serum albumin for 30 minutes at 37 °C. Sections were incubated overnight at 4 °C with the indicated primary antibodies diluted in 1% bovine serum albumin in PBS and Alex-488 secondary antibody (Invitrogen) for 1 h at 37 °C. DAPI (Sigma-Aldrich, St. Louis, MO) was used to stain the nucleus.
Rhodamine phalloidin (Cytoskeleton Inc., Denver, CO) staining was used to show actin rearrangement as indicated by the manufacturer’s instructions. In brief, the cells that reached semi confluence were stained for 30 min with 100 nM rhodamine phalloidin following fixation (4% paraformaldehyde) and permeabilization (0.1% Triton X-100). The cells were then counter stained with DAPI to highlight the position of the nuclei. Images were analyzed and collected with a Zeiss LSM 510 Confocal Imaging System (Zeiss, Jena, Germany).
Statistical analysis
All values were expressed as means ± SD. Student’s t-test and a one-way analysis of variance were used to analyze the differences among various groups. All probabilities were two-tailed, and p < 0.05 was considered statistically significant.
Results
FSGS reduced podocyte survival, WT1 level and raised miR-206 expression
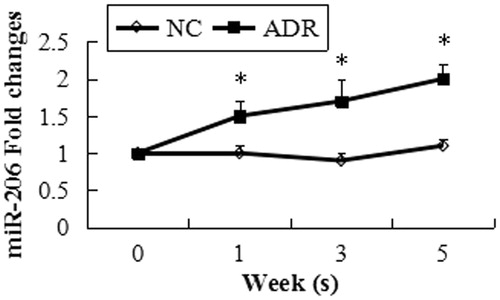
We utilized a mouse model of ADR-induced nephropathy, a model of human FSGS and widely recognized of podocyte injury to investigate the role of miR-206 in podocyte injury. We first analyzed glomerular pathology in ADR mice during the study period. ADR mice developed interstitial fibrosis and proliferative glomerular lesions during disease progression. Foot process effacement, basement membrane thickening were detected in the ADR mice by transmission electron microscopy (). In the ADR group, urinary protein excretion was significantly increased throughout the study period () and the kidney strongly displayed the podocyte injury marker desmin (). Few podocyte in FSGS kidneys weakly expressed podocyte markers WT1 and synaptopodin through the study period compared with the normal control kidneys (). Following the administration of ADR, the miR-206 expression level increased significantly in the mice glomeruli ().
Figure 1. Glomerular pathology and podocyte integrity of control and ADR mice. PAS staining (upper panels) and Masson staining (middle panels) showed ADR mice developed glomerular and interstitial fibrosis. Original magnification, ×200. Electron microscopy (lower panels) indicated that the podocyte foot processes lost their normal interdigitating pattern and showed effacement instead in ADR mice (TEM × 13500).
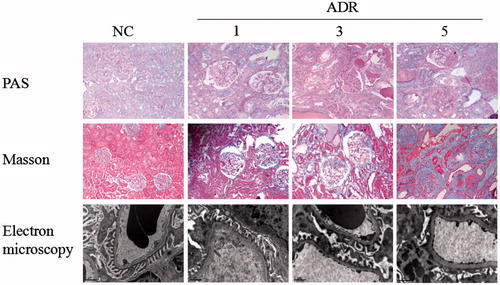
Figure 2. Level of urinary protein. Urinary protein excretion was significantly increased throughout the study period. Values = mean ± SD. A significant difference from the control value is indicated by *p < 0.05.
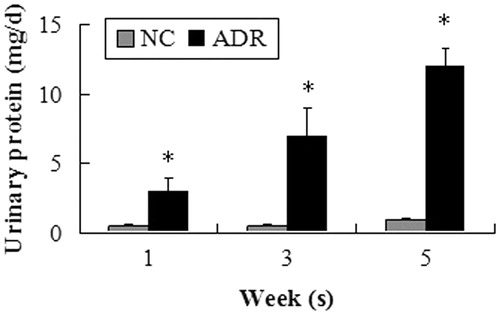
Figure 3. Expression of desmin. Podocyte in the ADR group strongly displayed the podocyte injury marker desmin. The intensities of the protein bands were quantified and normalized to β-actin. A significant difference from the control value is indicated by *p < 0.05.
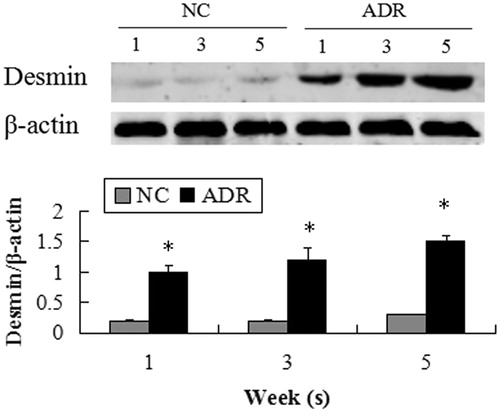
Figure 4. Expression of synaptopodin and WT1. FSGS kidneys weakly expressed cell markers synaptopodin and WT1 through the study period compared with the normal control kidneys.
Figure 5. miR-206 expression in kidneys of ADR mice. miR-206 was up-regulated in the ADR mice during the study period. Data are expressed as the mean ± SD calculated from six mice at each time point. *p < 0.05, significant difference from the normal control group.
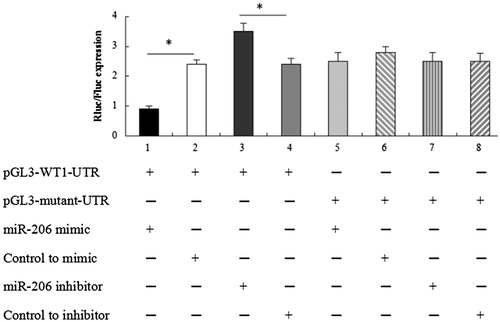
The 3'UTR of WT1 is directly targeted by miR-206
To determine whether WT1 was directly targeted by miR-206 as predicted by computer modeling, we constructed luciferase reporter plasmids that contained the 3'UTR of WT1 and generated mutations within the putative miR-206 target site. Using contransfection assays, we found that the activity of the 3'UTR of WT1 was suppressed by an miR-206 mimic, but not by a scrambled miRNA sequence ().
Figure 6. Dual-Luciferase reporter assays. The change in the Rluc/Fluc ratio indicates that the 3'UTR of the WT1 gene was directly targeted by miR-206. *p < 0.05.
Over-expression of miR-206 leads to podocyte injury
Forty-eight hours after transfecting miR-206 mimics and miRNA mimic negative control (miR-NC) into the MPC5 cells, the protein expression levels of markers related to podocyte injury were measured by western blot analysis. The expression of desmin significantly increased in the cells transfected with miR-206 mimics compared with the miR-NC group. However, the expressions of the miR-206 target WT1, as well as the podocyte functional marker synaptopodin were suppressed by transfection with miR-206 mimics (). Furthermore, the mRNA expressions of these genes were consistent with those of the protein expression ().
Figure 7. miR-206 promote podocyte damage. The expression of desmin significantly increased, while WT1 and synaptopodin were suppressed by transfection with miR-206 mimics compared with the miR-NC group. The intensities of the protein bands were quantified and normalized to β-actin. *p < 0.05 indicate statistically significant compared with the miR-NC group.
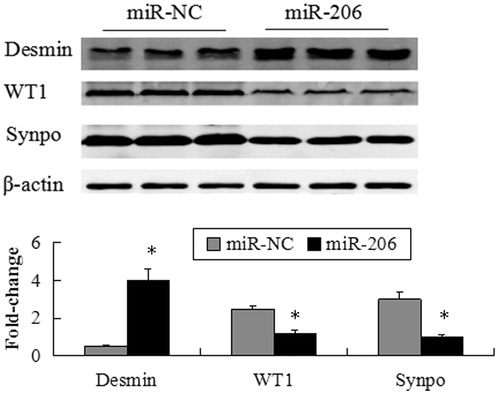
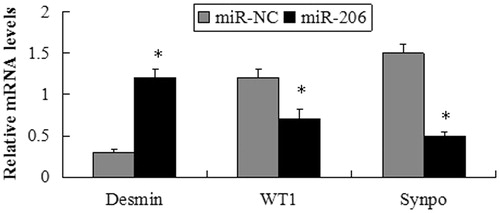
Figure 8. mRNA expression of desmin, WT1 and synaptopodin following transfection of the MPC5 cells with miR-206 mimics or miR-NC. The mRNA expression of desmin significantly increased, while WT1 and synaptopodin were suppressed by transfection with miR-206 mimics compared with the miR-NC group. *p < 0.05 indicate statistically significant compared with the miR-NC group.
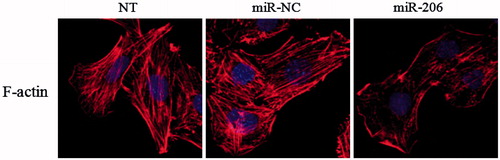
After transfection with miR-206 mimics, the podocyte cytoskeleton was disorder and rearrangement which may result in podocyte dysfunction and injury. The result revealed that miR-206 promoted the rearrangement of stress fiers in podocytes, causing them to localize around the cytomembrane compared with the control-transfected and untreated groups (NT) ().
Figure 9. Effects of miR-206 on cytoskeletal stabilization in podocyte. miR-206 promoted the rearrangement of stress fibers in podocyte, causing podocyte cytoskeletal injury compared with the NT and miR-NC group.
Discussion
FSGS is a major clinical problem because it does not respond to steroid therapy and leads to chronic renal failure. Despite recent advances, the cause of the majority of cases of FSGS remains elusive.Citation13 Podocyte loss and dysfunction in renal microenvironments are important features in the pathogenesis of FSGS.Citation2 Podocyte is a unique population of terminally-differentiated glomerular visceral epithelial cell that possess distinctive morphologic features exemplified by sophisticated foot processes.Citation14,Citation15 Under pathologic conditions, injurious stimuli often cause podocyte dedifferentiation characterized by loss of podocyte-specific features, thereby leading to podocyte dysfunction and defective glomerular filtration.Citation16,Citation17 miRNAs play a key role in podocyte injury.Citation18 It was previously shown that global podocyte-specific ablation of miRNAs by knockout of the miRNA processing enzymes Dicer or Drosha leads to proteinuria and glomerulosclerosis.Citation19,Citation20 Here, we provide a mechanistic insight into the molecular pathogenesis of podocyte injury induced by dysregulation of a single miRNA.
In the kidneys of mice treated with ADR, miR-206 was significantly upregulated, the mice had worse podocyte damage, glomerular fibrosis and renal dysfunction than those of the control mice, suggesting that harmful effects triggered by miR-206 signaling occurred in renal microenvironments. Using luciferase reporter assays, our results indicate that the prime target of miR-206 in podocyte is WT1. WT1, a gene that is essential for the development and maintenance of normal podocytes and glomeruli, is the predominant force that drives podocyte differentiation by promoting podocyte-specific gene expression.Citation3,Citation4 In humans, genetic defects in WT1 cause the Denys-Drash and Frasier syndromes, which are associated with the effaced podocyte, similarly to in FSGS.Citation13,Citation21 On the basis of these findings, we propose that the balance between miR-206 and WT1 is a key determinant that dictates the state of podocyte health and disease. In vitro, the cultured podocyte treated with ADR also supported the postulation that miR-206 signaling was destructive for maintaining podocyte homeostasis and that this is the initial pathogenic event in our model of FSGS. The present study demonstrates that the ectopic expression of miR-206 suppresses WT1 expression and results in podocyte injury.
Thus far, a great deal of understanding has focused on podocyte biology, particularly on the regulation of the actin cytoskeleton. As the main cytoskeletal component of podocyte processes, actin is the molecule ultimately responsible for podocyte shape and function. The changes commonly observed in podocyte damage, i.e. loss of stress fires, cell rounding and shortening of ramifications in vitro, foot process effacement and microvillous transformation in vivo, are determined by alterations of actin dynamics.Citation22 In the present study, we found that miR-206 could disrupt the location of the actin skeleton and is the detrimental factors for the stabilization of the podocyte cytoskeleton.
Taken together, in this study, we demonstrated that miR-206 was upregulated in models of podocyte injury, which can initiate a cascade of podocyte-destabilizing molecular events starting with the down-regulation of WT1, followed by synaptopodin degradation, actin disruption, and lead to podocyte injury and renal dysfunction. Our findings also shed light on the future study that miR-206 is a potential diagnostic biomarker for podocyte injury and that its inhibition may be a clinical therapeutic strategy for the podocytopathy.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Funding information
This study was funded by Guangdong Natural Science Foundation (grant number 2015A030313039).
Disclosure statement
The authors declare that they have no conflict of interest.
References
- Li M, Armelloni S, Zennaro C, et al. BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J Pathol. 2015;235:731–744.
- Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. 2015;11:76–87.
- Chau YY, Brownstein D, Mjoseng H, et al. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. Plos Genet. 2011;7:e1002404.
- Guo JK, Menke AL, Gubler MC, et al. WT1 is a key regulator of podocyte function: Reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet. 2002;11:651–659.
- Macconi D, Bonomelli M, Benigni A, et al. Pathophysiologic implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol. 2006;168:42–54.
- Jackson RJ, Standart N. How do microRNAs regulate gene expression? Sci STKE. 2007;36:e1.
- Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–263.
- Fogo AB. Animal models of FSGS: Lessons for pathogenesis and treatment. Semin Nephrol. 2003;23:161–171.
- Wang Y, Wang YP, Tay YC, Harris DC. Progressive Adriamycin nephropathy in mice: Sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–1804.
- Takemoto M, Asker N, Gerhardt H, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161:799–805.
- Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8:697–705.
- Giardino L, Armelloni S, Corbelli A, et al. Podocyte glutamatergic signaling contributes to the function of the glomerular filtration barrier. J Am Soc Nephrol. 2009;20:1929–1940.
- D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398–2411.
- Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307.
- Shankland SJ. The podocyte's response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147.
- Wiggins RC. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214.
- Kietzmann L, Guhr SS, Meyer TN, et al. MicroRNA-193a regulates the transdifferentiation of human parietal epithelial cells toward a podocyte phenotype. J Am Soc Nephrol. 2015;26:1389–1401.
- Shi S, Yu L, Chiu C, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19:2159–2169.
- Zhdanova O, Srivastava S, Di L, et al. The inducible deletion of Drosha and microRNAs in mature podocytes results in a collapsing glomerulopathy. Kidney Int. 2011;80:719–730.
- Ratelade J, Arrondel C, Hamard G, et al. A murine model of Denys-Drash syndrome reveals novel transcriptional targets of WT1 in podocytes. Hum Mol Genet. 2010;19:1–15.
- Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437.