Abstract
Introduction: Air pollution increases the risk of cardiovascular diseases. A proposed mechanism is that local airway inflammation leads to systemic inflammation, affecting coagulation and the long-term risk of atherosclerosis. One major source of air pollution is wood burning. Here we investigate whether exposure to two kinds of wood smoke, previously shown to cause airway effects, affects biomarkers of systemic inflammation, coagulation and lipid peroxidation.
Methods: Thirteen healthy adults were exposed to filtered air followed by two sessions of wood smoke for three hours, one week apart. One session used smoke from the start-up phase of the wood-burning cycle, and the other smoke from the burn-out phase. Mean particle mass concentrations were 295 µg/m3 and 146 µg/m3, and number concentrations were 140 000/cm3 and 100 000/cm3, respectively. Biomarkers were analyzed in samples of blood and urine taken before and several times after exposure. Results after wood smoke exposure were adjusted for exposure to filtered air.
Results: Markers of systemic inflammation and soluble adhesion molecules did not increase after wood smoke exposure. Effects on markers of coagulation were ambiguous, with minor decreases in fibrinogen and platelet counts and mixed results concerning the coagulation factors VII and VIII. Urinary F2-isoprostane, a consistent marker of in vivo lipid peroxidation, unexpectedly decreased after wood smoke exposure.
Conclusions: The effects on biomarkers of inflammation, coagulation and lipid peroxidation do not indicate an increased risk of cardiovascular diseases in healthy adults by short-term exposure to wood smoke at these moderate doses, previously shown to cause airway effects.
Introduction
Exposure to particulate air pollution increases short- and long-term cardiopulmonary morbidity and mortality (Brook et al., Citation2010; Pope & Dockery, Citation2006; Rückerl et al., Citation2011). Globally, exposure to particulate matter (PM) is an important and modifiable factor affecting public health, with an estimated 800 000 premature deaths per year caused by particles <2.5 µm (PM2.5) (Brook et al., Citation2010) and 1.6 million premature deaths per year caused by biomass and coal smoke (Smith & Maeuzahl-Feuz, Citation2004; WHO, Citation2006).
Half the world’s population use solid fuels indoor for cooking and heating (Rehfuess et al., Citation2006). Exposure levels can be very high, especially indoors in the developing world, but burning wood for residential heating is common in the developed world as well and can be a major source of PM (Naeher et al., Citation2007). In the European Union (EU-15), domestic wood stoves contributed 25% of annual PM2.5 emissions in the year 2000, an increase to 38% is expected by 2020 (Amann et al., Citation2005). Wintertime contributions can be much higher, with studies showing around 70% in northern Sweden (Hedberg et al., Citation2006), up to 90% in Seattle (Larson & Koenig, Citation1994) and more than 90% in Christchurch, New Zealand (McGowan et al., Citation2002).
Inhalation of PM causes pulmonary health effects, but the majority of adverse health effects involve the cardiovascular system. Researchers have suggested that PM may instigate cardiovascular health effects via three main pathways and these suggestions are supported by a number of studies. One suggestion is that PM induces airway oxidative stress and inflammation causing pulmonary cells to release systemic inflammatory and pro-thrombotic cytokines, leading to destabilization of plaques, increased risk of blood clotting and atherogenesis. Another suggestion is that a similar response may directly be caused by PM or particle constituents translocated into the systemic circulation. A third is that particle interactions with receptors in the lung may affect the autonomic nervous system, increasing the risk of arrhythmias (Brook, Citation2008; Brook et al., Citation2004, Citation2010; Mills et al., Citation2009).
However, health effects and mechanisms may vary between different kinds of PM. Air pollution is a complex heterogeneous mixture of particles of varying size, composition and origin. The toxicity is determined not only by weight and number but also by size, surface area and chemical composition, and the various components may interact synergistically or antagonistically (Brook et al., Citation2010; Donaldson & Tran, Citation2002; Schwarze et al., Citation2006). Specific studies of types of PM or emission source are therefore needed to determine their health effects.
Wood smoke is not as extensively studied as other major pollutants, but many studies have shown associations between it and respiratory health effects. The evidence available regarding cardiovascular outcomes is too limited to judge whether wood smoke particles are less toxic than other combustion-derived particles, and further experimental studies of acute effects of major types of biomass smoke have been called for (Naeher et al., Citation2007).
We therefore designed a study to compare the effects in healthy human subjects exposed to two different kinds of wood smoke PM, one from the start-up phase and one from the burn-out phase of the wood-burning cycle. In a previous paper, we reported effects on biomarkers of airway effects (increases of Clara Cell protein 16 in serum and urine and fraction of exhaled nitric oxide) (Stockfelt et al., Citation2012). These effects were similar to those in our first experimental wood smoke exposure study (Barregard et al., Citation2008), which used the same experimental chamber but higher doses of wood smoke PM. That study also showed effects on biomarkers of systemic inflammation, coagulation and lipid peroxidation (factor VIII, serum amyloid A (SAA) and 8-iso-prostaglandin F2α) (Barregard et al., Citation2006), which could indicate increased risks of cardiovascular disease (CVD) by short-term exposure to wood smoke.
In this study, we examine whether exposure-related effects on biomarkers of systemic inflammation, coagulation and lipid peroxidation in healthy subjects could be detected at these lower doses of wood smoke exposure, and whether there is a difference between wood smoke from the start-up phase and the burn-out phase of the combustion cycle.
Methods
Subjects
We recruited 16 healthy never-smokers (8 men and 8 women) aged 20–57 years of age (mean age 31) from among the staff of our department and students. All had normal spirometry values and none had symptomatic allergy. The subjects were not allowed to take any medication during the two days preceding each exposure session and had to be free from any known infections for at least one week prior to the session. Three subjects were excluded before data analysis because upper airway infections emerged just before or between the sessions, and thus all results presented below are based on 13 individuals (6 men and 7 women aged 20–57, mean age 34 years). The study was approved by the Regional Ethics Review Board of Gothenburg, and the subjects gave their written informed consent.
Experimental design
The design of the current study was similar to our previous wood smoke exposure study (Barregard et al., Citation2006, Citation2008; Sallsten et al., Citation2006), but differed in using (1) wood smoke from two different phases of the combustion cycle and (2) lower inhaled doses. Because the exposure time had been decreased from four to three hours and no periods of exercise were included (compared to 50 min), the volume of inhaled air was only about half that of the previous study.
The subjects were first exposed to filtered indoor air for three hours; one week later they were exposed to wood smoke from the start-up phase of the wood-burning cycle and another week later to wood smoke from the burn-out phase of the wood-burning cycle. The sessions were identical apart from the wood smoke exposure, and each started with blood and urine sampling before the subjects entered the exposure chamber. After exposure, samples were taken several times (). The subjects had free intake of soft drinks and water and a small snack in the middle of the exposure, but did not eat during the hour before the first blood sample was taken. For more details, see Stockfelt et al. (Citation2012).
Table 1. Timing of breath, blood and urine sampling in the first subject. A new subject started the schedule every 10 min.
Exposure chamber and generation of wood smoke
A more detailed description of the exposure chamber and the principles of wood smoke generation has been reported (Sallsten et al., Citation2006). Briefly, the chamber was 29 m3 with walls covered in teflon-impregnated glass-fiber fabric. Wood smoke was generated in a small cast-iron wood stove placed just outside the chamber and was mixed with filtered air to achieve the target concentration. A mixture of hardwood and softwood was used (50% birch, 50% spruce). A 2.5–3 kg batch of wood logs was ignited, and another batch of 1.5 kg was added approximately every 40 min.
In the session using wood smoke from the start-up phase, smoke was supplied to the chamber for 12–14 min, immediately after the new wood logs were added. In the session using smoke from the burn-out phase, smoke was supplied for 15 min, starting 25 min after wood was added. The PM2.5 mass concentration was controlled online to maintain the target concentration.
Sampling and characterization of wood smoke
PM2.5 mass concentrations, number concentrations and size distributions of particles, black carbon, trace elements, NO, NO2, CO, CO2, particulate polycyclic aromatic hydrocarbons (PAHs), some volatile organic compounds (VOCs), temperature and relative humidity were measured in the center of the chamber. The complete results and further descriptions of sampling technique and apparatus have been described previously (Sallsten et al., Citation2006, Stockfelt et al., Citation2012). The most important data are summarized in .
Table 2. Time weighted averages of the exposure measurements in all sessions. More details are presented in Stockfelt et al. (Citation2012).
Blood and urine sampling
Blood was collected by venipuncture in SST tubes for serum, citrate tubes for plasma and EDTA tubes for leukocyte counts. Leukocyte counts were performed on the same day, while the serum and plasma aliquots were stored frozen in polyethylene cryotubes until analysis. The urine samples were collected in polypropylene bottles at different time points, and the volumes were registered and aliquots stored frozen until analysis.
Biochemical analyses
Blood counts were performed with flow cytometry (Celldyn 400, Abbott, reagents from Abbott, Abbot Park, IL). High-sensitivity serum C-reactive protein (CRP) was measured by immunoturbidometry using modular equipment and reagents from Roche Diagnostics (Mannheim, Germany). The STA-R equipment and reagents from Diagnostica Stago (Asniéres, France) were used to determine fibrinogen in plasma based on the coagulation time at high thrombin concentration, factor VII (FVII) using a one-stage thromboplastin method, and factor VIIIc (FVIII) using a one-stage clotting measurement. d-dimers were measured using a sandwich enzyme-linked immunosorbent assay (ELISA) with reagents from Diagnostica Stago, Triolab AB (Molndal, Sweden). Von Willebrand factor (vWf) antigen was determined with a sandwich ELISA using antibodies from DakoCytomation (Solna, Sweden). SAA, soluble intercellular adhesion molecule-1 (sCAM-1), soluble vascular cell adhesion molecule-1 (sVCAM-1), soluble P-selectin (sP-selectin), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) were analyzed using commercial ELISA kits, for SAA from Anogen (Ontario, Canada) and the others R&D Quantikine (Abingdon, UK). 8-Iso-prostaglandin F2α (8-iso-PGF2α) was analyzed in urine using a radioimmunoassay as developed by Basu (Citation1998). Urinary 8-iso-PGF2α values are presented after adjustment for creatinine levels in urine.
Statistics
For all biomarkers, intra-individual differences at each time point were calculated by subtracting the changes after exposure to filtered air from the changes after exposure to each of the two wood smoke sessions, making each individual his or her own control. Statistical significance was tested with two-sided Student’s t test for normally distributed biomarkers (FVII, FVIII, vWf, f8/vWf, leukocytes, sP-selectin, platelets) and Wilcoxon’s signed rank test for those not normally distributed (CRP, fibrinogen, SAA, 8-iso-PGF2α, sICAM-1, sVCAM-1, d-dimer, IL-6, TNF-α). Point estimates of all significant differences are also presented, expressed as the median of the intra-individual differences divided by each individual’s baseline value before wood smoke exposure, as a percentage. Associations between biomarkers were assessed using Spearman’s rank correlation coefficient (rs), separately for each sample time. Biomarker levels below the detection limit were replaced with the detection limit divided by the square root of 2 (Hornung & Reed, Citation1990). Statistical calculations were made using SAS 9.2.
Results
Exposure characterization
The mean PM mass concentration was below the detection limit in the filtered air session, was 295 µg/m3 in the start-up session, and was 146 µg/m3 in the burn-out session (). Average PM number concentrations were 2900/cm3 in the filtered air session and 140 000/cm3 and 100 000/cm3 in the start-up and burn-out sessions, respectively. Nearly all particles were <1 µm, with 55%, 68% and 40% respectively, being ultrafine (<100 nm, PM0.1). The geometric mean diameter was 38 nm (σg = 1.7) in the start-up session and 83 nm (σg = 1.5) in the burn-out session.
The concentrations of particulate PAHs were higher in the start-up session than in the burn-out session (), and in both wood smoke sessions much higher than in the filtered air session. The concentrations of benzene, 1,3-butadiene, formaldehyde, acetaldehyde and naphthalene were also higher in the wood smoke sessions. The mean NO2 concentration was 0.01 ppm in the filtered air session and 0.03 ppm and 0.05 ppm in the start-up and burn-out sessions ().
Biomarkers of systemic effects
The main biomarkers of systemic inflammation did not increase after exposure to wood smoke compared to exposure to filtered air; the levels of IL-6, TNF-α and SAA did not change significantly after exposure at any sampling time (). The white blood cell (WBC) count did not change significantly either; but did show a clear diurnal variation with higher levels in afternoons than in the mornings. CRP was slightly lower four hours after exposure in the start-up session compared to exposure to filtered air (median difference −5%, p = 0.04).
Table 3. Biomarkers of systemic inflammation, coagulation and lipid peroxidation. Medians (ranges) in serum, plasma and urine in the filtered air session and the two sessions of wood smoke exposure at all sample times. For timing of samples see , N = 13.
The sCAMs did not increase after wood smoke exposure. sP-selectin did not change significantly at any sampling time. Neither did soluble ICAM-1 apart from an isolated minor net increase (+7%, p = 0.008) 47 h after exposure in the burn-out session due to a decrease after filtered air. sVCAM-1 was marginally decreased four hours after exposure in both wood smoke sessions compared to exposure to filtered air (−2% and −5%, p = 0.01 and p = 0.02, respectively).
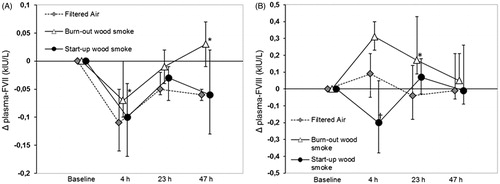
The effects of wood smoke on biomarkers of hemostasis were ambiguous. Fibrinogen decreased in the morning after exposure in both the start-up session (−4%, p = 0.04) and the burn-out session (−5%, p = 0.02) compared to exposure to filtered air. In the burn-out session, it was also lower four hours after exposure (−8%, p = 0.04, ). Platelet counts were also slightly lower four hours after exposure in the burn-out session (−6%, p = 0.003) and in the morning after exposure in both the start-up and burn-out sessions (−6% and −6%, p = 0.03 and 0.01, respectively). d-dimer showed a significant net decrease two mornings after exposure in the start-up session (−20%, p = 0.04). In the burn-out session, FVII showed a slight but significant increase compared to filtered air four hours after exposure, as well as two mornings after exposure (point estimates +6% and +12%, p = 0.006 and 0.04, respectively, ). In the same session, FVIII showed a non-significant tendency to increase four hours after wood smoke exposure (+14%, p = 0.09, ), and was significantly increased the next morning (+37%, p = 0.02), as was the FVIII/vWf ratio (+47%, p = 0.02). In the start-up session, FVIII also showed a tendency to increase the morning after exposure (+13%, p = 0.08), but had significantly decreased four hours after exposure (−29%, p = 0.03, ).
Figure 1. Median changes (Δ) from baseline and 90% CI for fibrinogen in plasma at all sample times in the filtered air session and both wood smoke sessions. †Significant net decrease after wood smoke exposure compared to after filtered air.
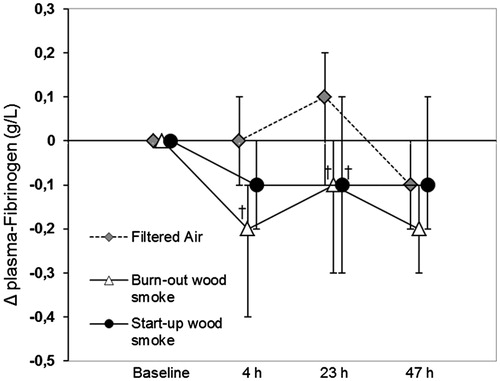
Figure 2. Median changes (Δ) from baseline and 90% CI for the coagulation factors VII (A) and VIII (B) at all sample times in the filtered air session and both wood smoke session. *Significantly net increase after wood smoke exposure; †Significant net decrease after wood smoke exposure compared to after filtered air.
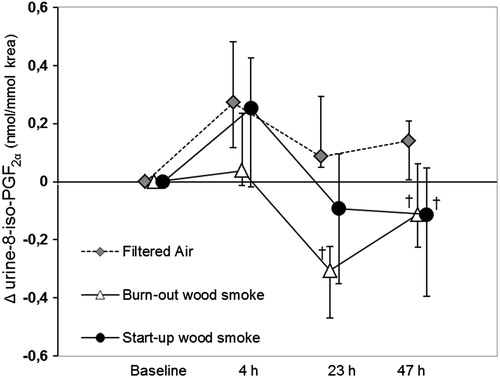
Concerning lipid peroxidation, the urinary isoprostane 8-iso-PGF2α adjusted for creatinine was markedly decreased after exposure to wood smoke; for the start-up session this occurred only the second morning after exposure (−53%, p = 0.02) and the burn-out session on the first and second mornings after exposure (−82% and −71%, p = 0.0002 and p = 0.006, respectively, ). This might in part be explained by an increase after exposure in the filtered air session. There was a diurnal variation with higher levels in the afternoons than in the mornings.
Figure 3. Median changes (Δ) from baseline and 90% CI for 8-iso-PGF2α in urine corrected for creatinine concentrations at all sample times in the filtered air session and both wood smoke sessions. †Significant net decrease after wood smoke exposure compared to after filtered air.
At most sample times, fibrinogen was highly correlated with both sP-selectin and leukocyte counts, CRP with SAA, FVIII with vWf and d-dimer, and FVII with platelet counts.
Discussion
This is the first study of systemic effects in humans of exposure to two kinds of wood smoke PM in the same experimental setup. Our a priori hypothesis was that this exposure would increase biomarkers of systemic inflammation, coagulation, and lipid peroxidation, thus indicating an increased risk of CVD. However, we found no consistent increases in any systemic biomarkers after exposure to smoke either from the start-up phase or the burn-out phase of the wood-burning cycle as compared to exposure to filtered air. As reported previously (Stockfelt et al., Citation2012), wood smoke at these exposure doses affected biomarkers of airway effects. In a previous study in the same exposure chamber with higher doses of wood smoke, we observed increases in biomarkers of both airway and systemic effects (Barregard et al., Citation2006, Citation2008). Taken together, these results indicate that local airway inflammation precedes systemic inflammation and has a lower threshold.
Markers of systemic inflammation and sCAMs
IL-6 and TNF-α are inflammatory cytokines that induce the production of acute phase reactants in the liver; among these reactants are CRP and SAA. They all increase in serum during inflammation, as does WBC count. WBCs are recruited from the blood to the site of inflammation with the aid of CAMs, the soluble forms of which are usually elevated in serum during inflammation. Inflammation plays a key role in the development of CVD. Of all the biomarkers of inflammation we measured, CRP is perhaps the best established predictor of CVD, but associations between increased serum levels and CVD/mortality have been found also for SAA, WBC, IL-6, TNF-α, sICAM-1, sVCAM-1 and sP-selectin (Harris et al., Citation1999; Huo & Ley, Citation2001; Libby, Citation2002; Pai et al., Citation2004; Pearson et al., Citation2003; Ridker et al., Citation2000; The Emerging Risk Factors Collaboration: Kaptoge, Citation2010, Citation2012).
For most biomarkers of systemic inflammation and soluble adhesion molecules, we found no significant changes after either of the wood smoke exposure session compared to exposure to filtered air (IL-6, TNF-α, SAA, WBC, sP-selectin), or only small sporadic changes, most likely explained by chance (CRP, sICAM-1, sVCAM-1). However, our first wood smoke exposure study, which used higher exposure doses found an increase in SAA and a decrease in IL-6 (Barregard et al., Citation2006). This can be interpreted as a dose-dependent short-term effect in healthy humans. Surprisingly, few other wood smoke exposure studies have published any results about systemic effects on biomarkers of inflammation and soluble adhesion molecules. One study (PM2.5 about 180 µg/m3) found an increased percentage of neutrophils but no increases in total WBC, IL-6 or TNF-α (Ghio et al., Citation2011); another study examining markers of inflammation and oxidative stress in peripheral blood monocytes found no increases in either CAMs or inflammatory cytokines (Forchhammer et al., Citation2012). In summary, moderate short-term exposure to wood smoke seems to have little effect on systemic inflammatory biomarkers. However, a recent real-world intervention study using indoor air filtration for one week in homes in a wood-burning community found decreases in CRP and IL-6 (Allen et al., Citation2011).
Biomarkers of coagulation and fibrinolysis
Inflammation and thrombosis are closely linked. Fibrinogen is an acute phase reactant that is increased in inflammatory conditions and an important determinant of blood viscosity and platelet aggregation. Plasma levels have been associated with CVDs (Fibrinogen Studies Collaboration: Danesh et al., 2005; Pearson et al., Citation2003; Stec et al., Citation2000; The Emerging Risk Factors Collaboration: Kaptoge, Citation2012). Platelets are important in both inflammation and thrombosis and are linked to the progression of atherosclerosis (Gawaz et al., Citation2005). Circulating levels of d-dimer reflects the degradation of cross-linked fibrin, and higher levels have been associated with coronary heart disease (Danesh et al., Citation2001).
Fibrinogen and platelet counts had both slightly but significantly decreased after exposure to wood smoke compared to exposure to filtered air at three sampling times. The few other wood smoke exposure studies investigating these biomarkers did not observe any effects, even though their exposure doses were higher (Barregard et al., Citation2006; Ghio et al., Citation2011). Studies of other air pollution PM sources have reported mixed results (Frampton et al., Citation2004; Ghio et al., Citation2000, Citation2003; Gong et al., Citation2003; Steinvil et al., Citation2008).
The coagulations factor VII and VIII and the vWf are part of the coagulation cascade. Factor VIII and vWf have been associated with inflammation and CVD (Folsom et al., Citation1997; Haverkate, Citation2002; Lacroix-Desmazes et al., Citation2008). The vWf was not affected in either of these studies nor in the only other study investigating it (Ghio et al., Citation2011). The coagulation factor VII was significantly increased at two sample times and factor VIII at one sample time after exposure to burn-out smoke compared with exposure to filtered air. However, the increase for factor VII was slight and was observed after four hours and after 47 hours but not in between these sampling times; and in the start-up session factor VIII instead decreased four hours after exposure. The only other published experimental wood smoke exposure study analyzing these coagulation factors observed an increase in factor VIII but not in FVII the morning after exposure, but the inhaled dose in that study was about twice as high (Barregard et al., Citation2006).
In summary, the effects on coagulation and fibrinolysis are unclear. While certain biomarkers increased at some time points after exposure to wood smoke, the increases were not consistent and were accompanied by a higher number of decreases. After putting these together, and taking other studies of wood smoke exposure into account, we do not interpret these results as a sign of increased risk of thrombosis in healthy adults after short-term exposure to moderate doses of wood smoke. Such effects may, however, occur at higher or longer exposures. Another possible interpretation of the results is that the decrease in fibrinogen and platelets represents a true effect by wood smoke exposure. Though slight, these effects were consistent for (the same) three sampling times for both biomarkers. A panel study that found a negative correlation between PM10 exposure and both fibrinogen and platelets speculated that this might represent a consumption of these factors as a response to nanoparticles (Seaton et al., Citation1999). However, the majority of studies find either a positive or null association between fibrinogen and particle exposure (Rückerl et al., Citation2011).
Lipid peroxidation
8-Iso-PGF2α, a major F2-isoprostane in urine, is considered to be a reliable marker of lipid peroxidation and oxidative stress in vivo. Cigarette smoking and CVDs are associated with elevated levels of F2-isoprostanes (Basu, Citation2008). Increases in markers of oxidative stress have been reported in a large number of in vitro and animal air pollution studies (Risom et al., Citation2005). Our hypothesis was therefore that 8-iso-PGF2α in urine (adjusted for creatinine or measured as excretion rate per hour) would increase after exposure to wood smoke. Surprisingly, there was a clear decrease in 8-iso-PGF2α in urine after exposure to wood smoke, especially in the burn-out session. We cannot find any biologically plausible explanation for this finding. Increased 8-iso-PGF2α in urine has been reported in one human experimental wood smoke exposure study (Barregard et al., Citation2006). Another wood smoke study found no changes in 8-iso-PGF2α in exhaled breath condensate after exposure (Riddervold et al., Citation2012), but a study of concentrated ambient particles found an increase (Mills et al., Citation2008). There are also studies reporting no significant changes after diesel exposure (Allen et al., Citation2009) or indoor air filtration (Allen et al., Citation2011; Brauner et al., Citation2008).
Comparisons between experimental wood smoke exposure studies
It has previously been stated that experimental data of PM toxicity would benefit from increased comparability of studies (Schwarze et al., Citation2006), which seems especially true for studies of wood smoke. Emissions from burning wood vary considerably depending on the wood type, combustion appliance, combustion conditions and phase of the combustion cycle (Kocbach Bolling et al., Citation2009; McDonald et al., Citation2000, Naeher et al., Citation2007). Evidence on which characteristics are important for health effects is limited, but animal and in vitro studies indicate that the effects might be different for particles from different combustion conditions (Danielsen et al., Citation2010, Citation2011; Kocbach Bolling et al., Citation2009), and that the organic fraction might be most important for the biological effects (Kocbach Bolling et al., Citation2012). Considering this, it is not surprising that the results are not completely consistent for the published experimental human wood smoke chamber exposure studies (Barregard et al., Citation2006, Citation2008; Forchhammer et al., Citation2012; Ghio et al., Citation2011; Pope et al., Citation2011; Riddervold et al., Citation2011, Citation2012; Sehlstedt et al., Citation2010; Stockfelt et al., Citation2012). There is great heterogeneity in experimental setups, wood smoke generation, subjects, exposure doses and outcomes measured, making a comparative analysis tentative at best. Compared to the experimental studies of wood smoke exposure, studies of real-world exposure more consistently report clear effects on biomarkers (Allen et al., Citation2011; Hejl et al., Citation2013; Swiston et al., Citation2008; Tan et al., Citation2000; Van Eeden et al., Citation2001). This might be explained by higher doses of wood smoke in most of these studies – mainly because of longer times of exposure. In the studies of firefighters (Hejl et al., Citation2013; Swiston et al., Citation2008) the levels of PM were also very high.
It would be valuable if future studies on wood smoke were more comparable. Biomarkers that are risk factors for CVDs may be especially important to study, considering the vast burden of disease due to particulate air pollution, and the evidence that the effects of wood smoke exposure is less for cardiovascular outcomes than for respiratory effects (Naeher et al., Citation2007). Also, testing several kinds of wood smoke in the same setup may provide evidence on differences in toxicity and using different doses of the same type of wood smoke may increase the understanding of the dose–response relationships. Moreover, it is important that also negative results are published to provide a complete picture and to avoid publication bias.
Strengths and limitations
One strength of this study is that we mainly used biomarkers associated with clinically relevant adverse outcomes, such as cardiovascular morbidity and mortality. Another strength is that we used the same experimental setup for two different wood smoke exposures, as well as for a previous wood smoke exposure study, enabling comparisons between the studies. The experimental chamber is large enough to house all the subjects in the same chamber, decreasing exposure variability compared to exposing subjects on many different days. A limitation is that we did not use a randomized cross-over design due to time limitations, making it difficult to exclude carry-over effects or effects of learning with certainty. This seems biologically unlikely though as we did not see any clear effects on the biomarkers by exposure to either filtered air or wood smoke, and we do not see any evidence of this in the pre-exposure levels of biomarkers. Moreover, since the smell of wood smoke is obvious and difficult to mask, we could not do a blind study regarding exposure to filtered air or wood smoke; however, the subjects were unaware of wood-burning cycles. One possible reason for non-positive findings that must be considered is lack of power. Post hoc power calculations show that with 80% power (two-tailed) we can detect changes of about 8–15% of pre-exposure values, which we think is acceptable power. The fact that the biomarkers often changed in the direction opposed to the hypothesis also argues against a true but not detected effect.
We used a relatively straight-forward analysis of intraindividual differences in the outcome variables, adjusted for effects at filtered air. Other statistical methods to analyze the data might be considered, but do not seem to alter the results substantially. A repeated-measures analysis of variance using PROC MIXED in SAS comparing effects of wood smoke to filtered air showed similar non-positive results for most biomarkers, a few but conflicting significant effects on the coagulation factors VII and VIII, and a decrease in 8-iso-PGF2α.
Conclusions
The results indicate that there is no increase in systemic inflammation or coagulation in healthy adults at these doses of wood smoke exposure, which has previously been shown to cause airway effects. Instead a few biomarkers associated with coagulation and lipid peroxidation showed a tendency to decrease after wood smoke exposure.
Declaration of interest
The authors declare no conflicts of interest.
This study was funded by the Swedish Council for Working Life and Social Research (FAS), and by the Swedish Environmental Protection Agency through the Swedish Clean Air Research Program (SCARP).
Acknowledgments
The authors thank Lena Samuelsson, Sandra Johannesson, Peter Molnar, Bo Strandberg, and Kerstin Bergemalm-Rynell (all from our department) for data collection.
Related Research Data
References
- Allen J, Trenga CA, Peretz A, et al. (2009). Effect of diesel exhaust inhalation on antioxidant and oxidative stress responses in adults with metabolic syndrome. Inhal Toxicol 21:1061–7
- Allen RW, Carlsten C, Karlen B, et al. (2011). An air filter intervention study of endothelial function among healthy adults in a woodsmoke-impacted community. Am J Respir Crit Care Med 183:1222–30
- Amann M, Bertok I, Cofala J, et al. (2005). Baseline Scenarios for the Clean Air for Europe (CAFE) Programme: final report, Laxenburg, Austria
- Barregard L, Sallsten G, Andersson L, et al. (2008). Experimental exposure to wood smoke: effects on airway inflammation and oxidative stress. Occup Environ Med 65:319–24
- Barregard L, Sallsten G, Gustafson P, et al. (2006). Experimental exposure to wood-smoke particles in healthy humans: effects on markers of inflammation, coagulation, and lipid peroxidation. Inhal Toxicol 18:845–53
- Basu S. (1998). Radioimmunoassay of 15-keto-13,14-dihydro-prostaglandin F2[alpha]: an index for inflammation via cyclooxygenase catalysed lipid peroxidation. Prostaglandins Leukot Essent Fatty Acids 58:347–52
- Basu S. (2008). F2-isoprostanes in human health and diseases: from molecular mechanisms to clinical implications. Antioxid Redox Signal 10:1405–34
- Brauner EV, Forchhammer L, Moller P, et al. (2008). Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med 177:419–25
- Brook RD. (2008). Cardiovascular effects of air pollution. Clin Sci 115:175–87
- Brook RD, Franklin B, Cascio W, et al. (2004). Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation 109:2655–71
- Brook RD, Rajagopalan S, Pope CA III, et al. (2010). Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation 121:2331–78
- Danesh J, Whincup P, Walker M, et al. (2001). Fibrin D-dimer and coronary heart disease. Circulation 103:2323–7
- Danielsen PH, Loft S, Jacobsen NR, et al. (2010). Oxidative stress, inflammation, and DNA damage in rats after intratracheal instillation or oral exposure to ambient air and wood smoke particulate matter. Toxicol Sci 118:574–85
- Danielsen PH, Møller P, Jensen KA, et al. (2011). Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem Res Toxicol 24:168–84
- Donaldson K, Tran CL. (2002). Inflammation caused by particles and fibers. Inhal Toxicol 14:5–27
- Danesh J, Lewington S, Thompson SG, et al; Fibrinogen Studies Collaboration. (2005). Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality. JAMA 294:1799–809
- Folsom AR, Wu KK, Rosamond WD, et al. (1997). Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation 96:1102–8
- Forchhammer L, Moller P, Riddervold I, et al. (2012). Controlled human wood smoke exposure: oxidative stress, inflammation and microvascular function. Part Fibre Toxicol 9:7
- Frampton MW UM, Zareba W, Oberdörster G, et al. (2004). Effects of exposure to ultrafine carbon particles in healthy humans and subjects with asthma. Research Reports -- Health Effects Institute 126:1–63
- Gawaz M, Langer H, May AE. (2005). Platelets in inflammation and atherogenesis. J Clin Invest 115:3378–84
- Ghio AJ, Hall A, Bassett MA, et al. (2003). Exposure to concentrated ambient air particles alters hematologic indices in humans. Inhal Toxicol 15:1465–78
- Ghio AJ, Kim C, Devlin RB. (2000). Concentrated ambient air particles induce mild pulmonary inflammation in healthy human volunteers. Am J Respir Crit Care Med 162:981–8
- Ghio AJ, Soukup JM, Case M, et al. (2011). Exposure to wood smoke particles produces inflammation in healthy volunteers. Occup Environ Med 69:170–5
- Gong H, Linn WS, Sioutas C, et al. (2003). Controlled exposures of healthy and asthmatic volunteers to concentrated ambient fine particles in Los Angeles. Inhal Toxicol 15:305–25
- Harris TB, Ferrucci L, Tracy RP, et al. (1999). Associations of elevated Interleukin-6 and C-Reactive protein levels with mortality in the elderly. Am J Med 106:506–12
- Haverkate F. (2002). Levels of haemostatic factors, arteriosclerosis and cardiovascular disease. Vasc Pharmacol 39:109–12
- Hedberg E, Johansson C, Johansson L, et al. (2006). Is levoglucosan a suitable quantitative tracer for wood burning? Comparison with receptor modeling on trace elements in Lycksele, Sweden. J Air Waste Manag Assoc 56:1669–78
- Hejl AM, Adetona O, Diaz-Sanchez D, et al. (2013). Inflammatory effects of woodsmoke exposure among wildland firefighters working at prescribed burns at the Savannah River Site, SC. J Occup Environ Hyg 10:173–80
- Hornung RW, Reed LD. (1990). Estimation of average concentration in the presence of non-detectable values. Appl Occup Environ Hyg 5:48–51
- Huo Y, Ley K. (2001). Adhesion molecules and atherogenesis. Acta Physiol Scand 173:35–43
- Kaptoge S, Di Angelantonio E, Lowe G, et al; The Emerging Risk Factors Collaboration. (2010). C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. The Lancet 375:132–40
- Kaptoge S, Di Angelantonio E, Pennells L, et al; The Emerging Risk Factors Collaboration. (2012). C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med 367:1310–20
- Kocbach Bolling A, Pagels J, Yttri K, et al. (2009). Health effects of residential wood smoke particles: the importance of combustion conditions and physicochemical particle properties. Part Fibre Toxicol 6:29
- Kocbach Bolling A, Totlandsdal A, Sallsten G, et al. (2012). Wood smoke particles from different combustion phases induce similar pro-inflammatory effects in a co-culture of monocyte and pneumocyte cell lines. Part Fibre Toxicol 9:45
- Lacroix-Desmazes S, Navarrete A-M, André S, et al. (2008). Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood 112:240–9
- Larson TV, Koenig JQ. (1994). Wood smoke -- emissions and noncancer respiratory effects. Annu Rev Public Health 15:133–56
- Libby P. (2002). Inflammation in atherosclerosis. Nature 420:868–74
- McDonald JD, Zielinska B, Fujita EM, et al. (2000). Fine particle and gaseous emission rates from residential wood combustion. Environ Sci Technol 34:2080–91
- McGowan JA, Hider PN, Chacko E, Town GI. (2002). Particulate air pollution and hospital admissions in Christchurch, New Zealand. Aust N Z J Public Health 26:23–9
- Mills NL, Donaldson K, Hadoke PW, et al. (2009). Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med 6:36–44
- Mills NL, Robinson SD, Fokkens PHB, et al. (2008). Exposure to concentrated ambient particles does not affect vascular function in patients with coronary heart disease. Environ Health Perspect 116:709–15
- Naeher LP, Brauer M, Lipsett M, et al. (2007). Woodsmoke health effects: a review. Inhal Toxicol 19:67–106
- Pai JK, Pischon T, Ma J, et al. (2004). Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med 351:2599–610
- Pearson TA, Mensah GA, Alexander RW, et al. (2003). Markers of inflammation and cardiovascular disease. Circulation 107:499–511
- Pope CA, Dockery DW. (2006). Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc 56:709–42
- Pope CA, Hansen JC, Kuprov R, et al. (2011). Vascular function and short-term exposure to fine particulate air pollution. J Air Waste Manag Assoc 61:858–63
- Rehfuess E, Mehta S, Üstün A. (2006). Assessing household solid fuel use: multiple implications for the millennium development goals. Environ Health Perspect 114:373–8
- Riddervold I, Bonlokke J, Olin A-C, et al. (2012). Effects of wood smoke particles from wood-burning stoves on the respiratory health of atopic humans. Part Fibre Toxicol 9:12
- Riddervold IS, Bønløkke JH, Mølhave L, et al. (2011). Wood smoke in a controlled exposure experiment with human volunteers. Inhal Toxicol 23:277–88
- Ridker PM, Hennekens CH, Buring JE, Rifai N. (2000). C-Reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342:836–43
- Risom L, Møller P, Loft S. (2005). Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res Fund Mol Mech Mut 592:119–37
- Rückerl R, Schneider A, Breitner S, et al. (2011). Health effects of particulate air pollution: a review of epidemiological evidence. Inhal Toxicol 23:555–92
- Sallsten G, Gustafson P, Johansson L, et al. (2006). Experimental wood smoke exposure in humans. Inhal Toxicol 18:855–64
- Schwarze PE, Øvrevik J, Låg M, et al. (2006). Particulate matter properties and health effects: consistency of epidemiological and toxicological studies. Hum Exp Toxicol 25:559–79
- Seaton A, Soutar A, Crawford V, et al. (1999). Particulate air pollution and the blood. Thorax 54:1027–32
- Sehlstedt M, Dove R, Boman C, et al. (2010). Antioxidant airway responses following experimental exposure to wood smoke in man. Part Fibre Toxicol 7:21
- Smith KR MS, Maeuzahl-Feuz M. (2004). Indoor air pollution from household use of solid fuels. In: Ezzati M, Rodgers A, Lopez AD, Murray CJL. (eds.) Comparative quantification of health risks: global and regional burden of disease due to selected major risk factors. Chapter 18. Geneva: World Health Organization, 1435--93
- Stec JJ, Silbershatz H, Tofler GH, et al. (2000). Association of fibrinogen with cardiovascular risk factors and cardiovascular disease in the framingham offspring population. Circulation 102:1634–8
- Steinvil A, Kordova-Biezuner L, Shapira I, et al. (2008). Short-term exposure to air pollution and inflammation-sensitive biomarkers. Environ Res 106:51–61
- Stockfelt L, Sallsten G, Olin A-C, et al. (2012). Effects on airways of short-term exposure to two kinds of wood smoke in a chamber study of healthy humans. Inhal Toxicol 24:47–59
- Swiston JR, Davidson W, Attridge S, et al. (2008). Wood smoke exposure induces a pulmonary and systemic inflammatory response in firefighters. Eur Respir J 32:129–38
- Tan W, Qiu D, Liam B, et al. (2000). The human bone marrow response to acute air pollution caused by forest fires. Am J Respir Crit Care Med 161:1213–17
- Van Eeden S, Tan W, Suwa T, et al. (2001). Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM10). Am J Respir Crit Care Med 164:826–30
- WHO. (2006). Fuel for life: household energy and health. Geneva: World Health Organisation