Death receptor 3 (DR3, TRAMP, LARD, Apo3, Wsl1, TNFRSF25) is a member of the tumor necrosis factor receptor superfamily (TNFRSF) and shows closest homology to TNFR1.Citation1 Like TNFR1, DR3 contains 4 extracellular cysteine-rich repeats and is capable of signaling both apoptosis via caspase 8 activation and cell survival via the activation of NFκB.Citation2 In the immune system, DR3 has been shown to affect negative selection during thymocyte developmentCitation3 and can modulate T, NKT, and myeloid cell function. Essential roles for DR3 and its only confirmed ligand TNF-like protein 1A (TL1A) have been demonstrated in murine models of experimental autoimmune encephalomyelitis (EAE),Citation4 experimental colitis,Citation5 allergic lung inflammation,Citation4,Citation6 and inflammatory arthritis,Citation7 while the DR3/TL1A axis has been associated with a number of inflammatory diseases in humans, including rheumatoid arthritis.Citation8
Experimental autoimmune uveoretinitis (EAU) is an organ-specific, CD4+ T-cell-mediated disease that targets the retina.Citation9 Studies in EAU have provided abundant evidence that members of the tumor necrosis factor superfamily play pivotal roles in the pathogenesis of intraocular inflammation and retinal tissue destruction.Citation10–14 CD4+ Th1 cells directed toward retinal antigens, produce cytokines, such as IFNγ and TNFα, which activate resident and infiltrating mononuclear cells to generate inflammatory mediators such as nitric oxide (NO), essential for maximum tissue destruction. As DR3 is essential for the development of other autoimmune and inflammatory diseases, we tested it’s involvement in the pathogenesis of EAU.
EAU was induced in 6- to 8-week-old C57Bl/6 (DR3WT) or DR3-deficient (DR3KO, generated as previously described and supplied by CRUKCitation3) mice by immunization with a subcutaneous injection of 500 µg per mouse of interphotoreceptor retinoid-binding protein (IRBP) peptide residues 1–20 (GPTHLFQPSLVLDMAKVLLD; Sigma, Genosys, UK), emulsified in vol/vol complete Freund’s adjuvant (supplemented with 2.5 mg/mL Mycobacterium tuberculosisCitation12), with an additional intraperitoneal injection of 1.5 µg of Bordetella pertussis toxin (Sigma) on day 0. Animals were specific pathogen-free, isolator-reared, and maintained in accordance to Home Office Regulations for Animal Experimentation, UK. Mice were sacrificed by CO2 asphyxiation on day 21 postimmunization, normally the peak of disease in C57Bl/6 mice. Eyes were enucleated, sectioned, and stained with hematoxylin and eosin (H&E) for histological grading. The disease incidence in the DR3KO mice was significantly reduced (3 of 8) compared with DR3WT mice (6 of 6), implying partial resistance. Disease grading was determined by scoring for cellular infiltrate and structural changes (describedCitation12) and showed that DR3KO mice displayed a significantly reduced mean score of 0.5 ± 0.4 (maximum score 4) compared to age- and sex-matched DR3WT littermates (mean score of 3 ± 0.5; p = .03) (; data shown of two pooled independent experiments; Fisher’s exact test used to determine statistical significance).
FIGURE 1 187 X 250mm (72 x 72 DPI)
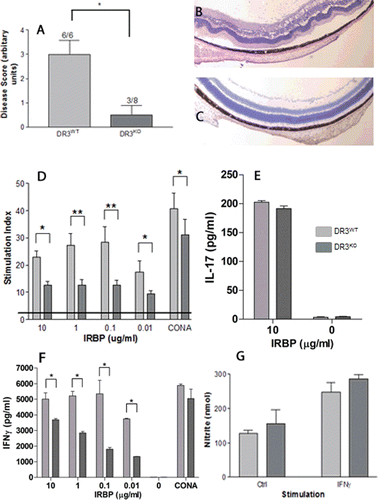
Splenocytes from DR3KO and DR3WT mice were assessed for proliferative ability to priming antigen at different time points following induction of EAU. Spleens were removed and splenocytes were extracted, seeded at 1 × 106/mL, and stimulated with a range of concentrations of IRBP1-20 (0.01–1 µg/mL) for 72 h. For the last 16 h, cells were pulsed with 0.5 µCi 3[H]thymidine (Amersham Biosciences) and uptake was measured using a beta counter (Wallac, Perkinelmer). PBS/2% DMSO and 2.5 µg/mL Con A (Concavalin A, Sigma) were used as negative and positive controls, respectively. Results were calculated as the geometric mean counts per minute (cpm) ± SEM of quadruplicate cultures and converted into a stimulation index (SI). The SI was calculated by dividing stimulation-induced proliferation by the negative control. There were no significant changes in proliferative ability of DR3KO and DR3WT splenocytes in response to IRBP1-20 10 days following EAU induction (data not shown). However, by day 21 (peak day of disease) significant differences between DR3WT and DR3KO IRBP1-20 stimulated splenocyte cultures were observed (; *p > .05 and **p > .005); DR3KO splenocytes demonstrated reduced peptide (IRBP1-20)-specific proliferation compared to their DR3WT counterparts. Concomitant production of IFNγ and IL-17 were measured by ELISA (OptEIA, Pharmingen, UK and R&D, UK, respectively). DR3KO splenocytes produced equivalent amounts of IL-17 (), but significantly reduced IFNγ () compared with DR3WT splenocytes. These data indicate that T-cell priming is intact in DR3KO mice, but sustained splenic T-cell responses seem impaired. The exact mechanisms underlying this phenomenon remain an area of further study.
Macrophages are believed the main mediator for tissue damage in EAU, because of their ability to cause peroxidation of the lipid membrane by the production of nitric oxide (NO). In the absence of TNFR1, macrophages are unable to produce NO in response to IFNγ, leading to minimal tissue destruction and resistance to EAU. As retinal destruction was markedly reduced in DR3KO mice, we wanted to determine whether DR3KO macrophage activation was impaired, as has been previously observed in TNFR1KO mice.Citation12 We generated bone marrow macrophages (BM-MØ) in Teflon bags as previously describedCitation12; these F4/80-positive macrophages were nonactivated and exhibited a resting phenotype (MHCIIlow, CD11blow, and CD86low; data not shown). Both DR3WT and DR3KO macrophages were able to produce NO in response to IFNγ stimulation (248 ± 29 and 287 ± 12 nM, respectively) compared with unstimulated controls (media alone; 127 ± 10 and 156 ±40 nM, respectively) ().
Overall, these results are completely consistent with previous studies of DR3KO mice in inflammatory diseases, although it is clear that DR3 function is very diverse, acting on stromal as well as immune cells. In renal inflammation, TL1A acts in a DR3-dependent fashion on tubular epithelial cells within kidney organ cultures, inducing NFκB and caspases with the potential of both promoting and protecting against injury.Citation15 In inflammatory arthritis,Citation7 DR3 helps promote the differentiation of macrophages to bone-damaging osteoclasts. We clearly show that intrinsic production of NO by macrophages in response to IFNγ is not impaired in DR3KO mice and this is unlikely to be the primary mechanism through which the absence of DR3 confers resistance to EAU. Instead, the impaired development of IFNγ-producing effector CD4+ T cells in the absence of DR3 is a more likely explanation, although the impact of DR3 signaling on other cytokines and chemokines cannot be ruled out. This would be in keeping with observations in allergic lung inflammation, neuroinflammation,Citation4 and colitis,Citation5 where DR3 drives the accumulation of effector T cells, whether they are Th1, Th2, or Th17.
Perhaps of greatest significance, all of our findings highlight the differences between DR3 and other members of the TNFR superfamily in the induction of ocular inflammation. Apart from the role of TNFR1 described above, Fas,Citation11 CD40L,Citation13 and 4-1BBLCitation14 have also been implicated in the pathogenesis of EAU. Mice deficient in 4-1BB (TNFRSF9) are susceptible to EAU, however, treatment with agonistic anti-4-1BB mAb alleviated disease through the expansion of IFNγ-producing CD11c+CD8+ T cells. IFNγ produced by these cells induces indoleamine 2,3-dioxygenase (IDO) in dendritic cells, and the induced IDO then becomes a suppressor of antigen-specific CD4+ T cells.Citation14 Disruption of CD40/CD40L (TNFRSF5/TNFSF5) interactions by administration of an anti-CD40L mAb completely protected against EAU and suppressed most related immunological responses, including DTH and cellular responses. CD40L blockade inhibited innate responses to immunization and reduced priming.Citation13 Fas (gld; TNFRSF6) or FasL (lpr) deficient mice developed lower incidence and severity following EAU induction compared with wild-type controls. The underlying mechanisms for this resistance were not defined, but bone marrow chimaeras demonstrated that normal expression of Fas and FasL on hemopoietic cells of the immune system, but not on ocular tissue, was required for the induction.Citation11
It is clear that although DR3 is the closest structural relative to TNFR1 and has similar signaling properties to other TNFRSF members, its biological functions are distinct and should represent an area of increasing interest in inflammatory research.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Kitson J, Raven T, Jiang YP et al. A death-domain-containing receptor that mediates apoptosis Nature 1996;384:372–375.
- Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J Biol Chem 2003;278:39251–39258.
- Wang EC, Thern A, Denzel A, Kitson J, Farrow SN, Owen MJ. DR3 regulates negative selection during thymocyte development. Mol Cell Biol 2001;21:3451–3461.
- Meylan F, Davidson TS, Kahle E et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 2008;29:79–89.
- Bamias G, Mishina M, Nyce M, Ross WG, Kollias G, Rivera-Nieves J, Pizarro TT, Cominelli F. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci U S A 2006;103:8441–8446.
- Fang L, Adkins B, Deyev V, Podack ER. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J Exp Med 2008;205:1037–1048.
- Bull MJ, Williams AS, MecklenburghZ, et al. The Death Receptor 3-TNF-like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J Exp Med. 2008;205:2457–2464.
- Bamias G, Siakavellas SI, Stamatelopoulos KS, Chryssochoou E, Papamichael C, Sfikakis PP. Circulating levels of TNF-like cytokine 1A (TL1A) and its decoy receptor 3 (DcR3) in rheumatoid arthritis. Clin Immunol 2008;129:249–255.
- Wacker WB, Lipton MM. Experimental allergic uveitis: homologous retina as uveitogenic antigen. Nature 1965;206:253–254.
- Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU). Prog Retin Eye Res 2004;23:617–637.
- Wahlsten JL, Gitchell HL, Chan C-C, Wiggert B, Caspi RR. Fas and Fas ligand expressed on cells of the immune system, not on the target tissue, control induction of experimental autoimmune uveitis. J Immunol 2000;165:5480–5486.
- Calder CJ, Nicholson LB, Dick AD. A selective role for the TNF p55 receptor in autocrine signaling following IFN-gamma stimulation in experimental autoimmune uveoretinitis. J Immunol 2005;175:6286–6293.
- Bagenstose LM, Agarwal RK, Silver PB et al. Disruption of CD40/CD40-ligand interactions in a retinal autoimmunity model results in protection without tolerance. J Immunol 2005;175:124–130.
- Choi BK, Asai T, Vinay DS, Kim YH, Kwon BS. 4-1BB-mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2,3-dioxygenase-dependent mechanisms. Cytokine 2006;34:233–242.
- Al-Lamki RS, Wang J, Tolkovsky AM, et al. TL1A both promotes and protects from renal inflammation and injury. J Am Soc Nephrol 2008;19:953–960.