Abstract
Purpose: We report an unusual case of orbital IgG4-related disease and discuss the distinguishing characteristics of the ophthalmic disease subtype.
Design: Case report.
Methods: Literature review and case description.
Results: Although lacrimal gland involvement has always been reported and elevated serum IgG4 is commonly observed, our case demonstrated neither in light of biopsy-proven IgG4 orbital involvement. A course of systemic steroids resolved our patient’s periorbital abnormalities.
Conclusions: IgG4-related orbital disease mandates a high index of suspicion, and should be confirmed by tissue biopsy. Possible progression to MALT lymphoma necessitates close surveillance and may require repeat biopsy.
A 23-year-old Caucasian female was referred from her primary care physician to the ophthalmology department for unresolved, painless blepharitis. The patient reported a dramatic increase in eyelid edema over the course of 6 weeks. She denied any history of contact lens wear, foreign body, or trauma. Furthermore, the patient's review of systems was negative for any recent weight loss, fever, or joint pain. On examination, a discrete, rubbery mass could be palpated in her right upper eyelid with associated blepharoptosis (marginal reflex distance-1 of 0.5 mm on the right relative to 4 mm on the left). The right eye demonstrated hypoglobus (2 mm relative to the left eye) and proptosis (Hertel’s exophthalmometry measured the right corneal surface position at 20 mm and the left at 17 mm, with a distance of 99 mm between the bilateral orbital rims) (). There was neither tenderness nor any limitation of extraocular motility. Visual acuity with correction was 20/25 in the right eye and 20/20 in the left. Computed tomography (CT) of the orbits revealed a lobulated, superior-temporal right orbital mass measuring 4.2 cm × 3.2 cm × 9 mm contiguous with the superior rectus muscle, without bony erosion (). Magnetic resonance imaging (MRI) confirmed mass extension medially reaching the trochlea and laterally abutting the lacrimal gland ().
Figure 1. External photograph on initial presentation showing right upper eyelid fullness, edema, and blepharoptosis.
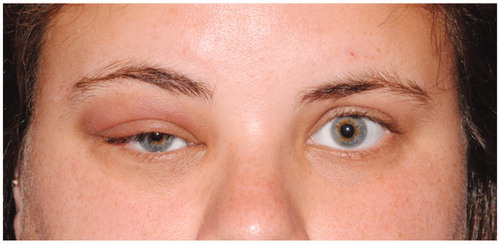
Figure 2. (A) Right globe proptosis with confluent edema within right extraconal supraorbital soft tissues, extending to posterior one-third of orbit. (B) CT orbit (soft tissue window), showing the mass occupying the right orbital superior space without bone remodeling.
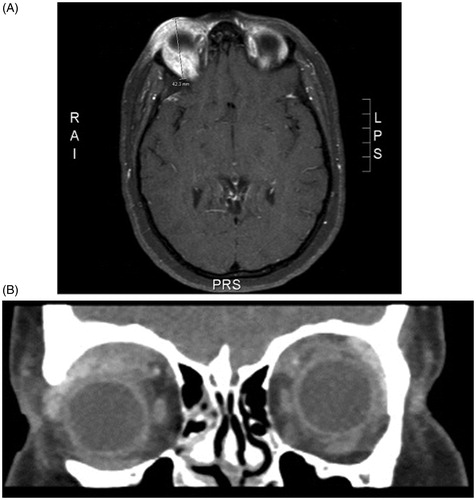
Figure 3. MRI (T1 STIR sequence), showing the mass in the right superior orbit displacing the globe inferiorly. The mass is seen extending from the trochlea to the lacrimal gland and abutting the superior rectus - levator muscle complex.
A differential diagnosis at this time included idiopathic orbital inflammation, Wegener granulomatosis, sarcoidosis or an orbital sarcoid reaction, IgG4-related sclerosing disease, Graves disease, Lyme disease, orbital tuberculosis, and syphilis. Other major concerns were lymphoma, a malignant tumor of the lacrimal gland, such as pleomorphic adenocarcinoma or adenoid cystic carcinoma, an orbital sarcoma, or metastatic disease from an unknown distant primary cancer.
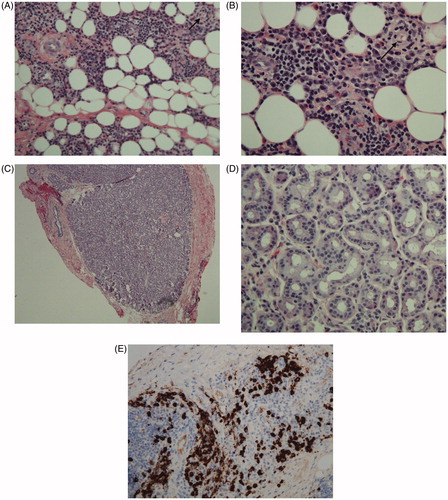
The patient was scheduled for a right anterior orbitotomy for exploration with open incisional biopsies of the mass and lacrimal gland. Histopathology revealed chronic inflammation, sclerosis, obliterative phlebitis, and IgG4-positive plasma cells (30–40% of all plasma cells; ∼50 cells per high-power field), diagnostic of IgG4 sclerosing disease (). Notably, the lacrimal gland was normal. Steroid therapy was initiated and the patient was referred to the rheumatology department for systemic evaluation. Subsequently, serology for Wegener granulomatosis (anti-neutrophil cytoplasmic antibodies, myeloperoxidase, proteinase 3), rheumatoid arthritis (anti-nuclear antibody, rheumatoid factor), sarcoidosis (angiotensin-converting enzyme, lysozyme), Graves disease (thyroid-stimulating immunoglobulin, thyroperoxidase), and Lyme disease (serum antibody) all returned negative. Inflammatory markers revealed an elevated C-reactive protein (6.03 mg/dL, normal value <1 mg/dL) but normal erythrocyte sedimentation rate. Additionally, a chest radiograph was unremarkable for lymphadenopathy. Thus, no underlying autoimmune etiology was uncovered. Her serum IgG4 level, which is characteristically elevated in systemic IgG4 disease, was normal.
Figure 4. (A) H&E stained slide of orbital mass showing orbital fat with fibrosis, marked plasma cell infiltrate, and obliterative phlebitis (indicated by arrow). (B) H&E stained slide of orbital mass showing orbital fat with fibrosis, marked plasma cell infiltrate, and obliterative phlebitis (indicated by arrow). (C) H&E stained slide of lacrimal gland showing normal architecture with no fibrosis or significant inflammation (examined under low power). (D) H&E stained slide of lacrimal gland showing normal architecture with no fibrosis or significant inflammation (examined under high power). (E) Immunohistochemical stain for IgG4 showing markedly increased number of IgG4 plasma cells.
IgG4-related disease is a relatively recently identified entity affecting multiple organs with a common histoseropathology. The landmark Hamano et al. publication first associated autoimmune pancreatitis with increased serum IgG4 levels, prompting the use of IgG4 as a serological marker of disease.Citation1 The authors also demonstrated an elevated number of IgG4-positive plasma cells within the affected tissue. These discoveries led to the recognition of various sclerosing disease patterns that today comprise the continuum of IgG4-related disease, including pachymeningitis, sclerosing sialadenitis, and inflammatory aortic aneurysm.
Orbital adnexal IgG4-related disease demonstrates uniform features, such as dense fibrosis, marked lymphoplasmacytic infiltration, and an abundance of IgG4-positive plasma cells.Citation2 Anatomically, bilateral lacrimal gland involvement with conjunctival sparing is commonly observed.Citation3 Obliterative phlebitis, although seen in systemic IgG4-related disease, is characteristically absent in orbital adnexal IgG4 sclerosing disease.Citation4 Once identified, ophthalmic IgG4 disease responds well to steroid therapy. Serum IgG4 levels, however, remain elevated long after disease remission.Citation5
Perhaps the most noteworthy distinction of orbital adnexal IgG4-sclerosing disease is the propensity for development of MALT lymphoma. Several studies have demonstrated this progression, otherwise rarely seen in disease affecting the pancreas and submandibular glands. Cheuk et al. estimate 10% of IgG4-related chronic sclerosing dacryoadenitis can progress into orbital lymphoma. It seems likely, therefore, that characteristics unique to the ophthalmic disease subtype predispose the orbit to lymphoma. Chronic hyperplasia of lymphoid tissue may lead to morphological changes and immunoglobulin gene rearrangement, resulting in B-cell proliferation and eventual malignant transformation.Citation5 Whether the lymphoma arises from neoplastic IgG4 cell populations or as a transformation of chronic IgG4-related background inflammation to tumor tissue remains unclear at this time.
In our patient, the orbital mass was composed of inflammatory tissue with a predominance of IgG4-positive plasma cells and evident phlebitis. Inflammation was absent from the lacrimal gland; moreover, the patient's serum IgG4 remained negative throughout her clinical course. As mentioned earlier, the lacrimal gland is characteristically affected by IgG4-related disease bilaterally and phlebitis is rarely observed in the ocular subtype. The unusual features in this patient challenge previously documented cases of orbital IgG4-related disease. However, per the most recently published diagnostic criteria, our case meets all three "histologically highly suggestive" features of IgG4-related disease.Citation6,Citation7 These include dense lymphoplasmacytic infiltrates, fibrosis (even though nonstoriform), and obliterative phlebitis. Most importantly, an elevated IgG4:IgG plasma cell ratio was present in the tissue. Although serum IgG4 was not increased, studies show that up to 40% of patients with biopsy-proven IgG4 disease do not demonstrate an increase in serum IgG4.Citation8
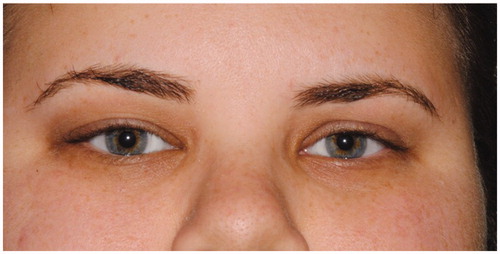
The patient responded very well to an oral course of steroids (prednisone 1 mg/kg, starting dose 80 mg daily) slowly tapered over 12 weeks (). Sixteen months following completion of treatment, the patient remains asymptomatic.
Figure 5. External photograph three months after the initial presentation showing total resolution of periorbital changes.
It should be duly noted that seemingly isolated orbital disease might have pronounced systemic sequelae. As with all clinical inquiries, it is imperative to form a complete differential diagnosis and to rule out the presence of any underlying systemic or autoimmune disease. Nonhealing cases of presumed blepharitis, hordeola, or chalazia by general medical or eye practitioners should perhaps be questioned and likely transferred to a specialist for a closer evaluation. In light of possible progression to MALT lymphoma, a diagnosis of IgG4-related disease mandates strong clinical suspicion and continued surveillance at regular follow-up visits. Although there is no standard protocol for follow-up of orbital IgG4-related disease, we recommend that, once asymptomatic, patients be seen at 3- to 6-month intervals until findings stabilize over a 1-year period. At that point, annual follow-up may be adequate. The prognosis seems favorable in the short-term with marked response to steroid therapy; however, the long-term implications for this entity are yet to be determined. Recurrent or new symptoms should be investigated promptly with clinical examination and imaging. Although MRI is preferable to CT for soft tissue detail, CT is more readily available and can show bony changes, including remodeling and erosion. In certain cases, it may even be necessary to perform a repeat biopsy to evaluate for malignant transformation if the response to steroids becomes suboptimal and the use of immune-modulators or radiation is contemplated. Consequently, it is important to coordinate the care of a patient with IgG4-related disease appropriately among the ophthalmologist, rheumatologist, and possibly an oncologist.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Hamano H, Kawa S, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–738
- Sato Y, Notohara K, et al. IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int 2010;60:247–258
- Mudhar HS, Bhatt R. Xanthogranulomatous variant of immunoglobulin G4 sclerosing disease presenting as ptosis, proptosis and eyelid skin plaques. Int Ophthalmol 2011;31:245–248
- Sato Y, Ohshima K, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008;58:465–470
- Cheuk W, Yuen HKL, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32:1159–1167
- Deshpande V, Zen Y, et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathol 2012;25:1181–1192
- Okazaki K, Umehara H. Review article: are classification criteria for IgG4-RD now possible? The concept of IgG4-related disease and proposal of comprehensive diagnostic criteria in Japan. Int J Rheumatol. 2012;2012:1–9
- Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011;23:108–113