
Abstract
Purpose: To report atypical ophthalmologic manifestations and complications of IgG4-related disease (IgG4-RD).
Methods: Patients with isolated ophthalmologic involvement of IgG4-RD other than lacrimal or orbital infiltration seen between 2009 and 2011 in a single tertiary center were retrospectively reviewed and their clinical and histological features, treatment, and prognosis were studied.
Case reports: Two patients (mean age 56.5 years) were included. One patient presented with recurrent anterior and posterior scleritis, and one patient had chronic conjunctival infiltration. Histopathology demonstrated lymphoplasmacytic proliferation with overexpression of IgG4+ plasma cells. Both patients initially responded to a high dose of oral corticosteroids (1 mg/kg/d). However, one patient required the adjunction of methotrexate and one patient developed an intra-epithelial conjunctival carcinoma on the site of the initial lesion.
Conclusion: Patients with atypical presentation of IgG4-RD, such as chronic conjunctival infiltration or scleritis, can suffer from considerable diagnostic delay leading to fibrosis or malignancy development. We report the first case of conjunctival carcinoma in a patient with IgG4-RD.
IgG4-related disease (IgG4-RD) is a recently defined clinical entity characterized by a multiorgan lymphoplasmacytic infiltrate with IgG4+ staining, associated with fibrosis or sclerosis. Initially described in 2001 by Hamano et al. in patients with autoimmune sclerosing pancreatitis and elevated IgG4 serum rate,Citation1 it was individualized by Neild et al.Citation2 in 2006 as hyper-IgG4 disease, regrouping several cases of multifocal sclerosing inflammatory diseases reported in the literature (multifocal sclerofibrosis, xanthofibrogranulomatosis, Ormond disease, etc). Several reviews have been publishedCitation3–5 leading to the most recent definition of this entity as IgG4-related disease. IgG-RD may involve multiple organs,Citation2–5 especially autoimmune sclerosing pancreatitis, retroperitoneal fibrosis, Reidel thyroiditis or hypothyroidism, sclerosing sialadenitis, and sclerosing cholangitis. The diagnostic criteriaCitation6 include diffuse or localized swelling or masses in a single or multiple organs, elevated serum IgG4 rate (>135 mg/dL), histopathologic lymphoplasmacytic infiltration with IgG4+/IgG+ cells ratio >40%, and >10 IgG4+ cells/HPF associated with storiform fibrosis and obliterative periphlebitis. Exclusion of alternative diagnoses, such as sarcoidosis, Castelman disease, Wegener vascularitis, lymphoma, or cancer, is also mandatory in order to state the diagnosis. Patients must also respond well to corticosteroids.Citation3,Citation7
Ophthalmic involvement typically presents as idiopathic orbital inflammation and chronic sclerosing dacryoadenitis.Citation8 We report atypical cases of IgG4-RD revealed by chronic relapsing scleritis and conjunctival infiltration, and report the first case of intraepithelial conjunctival carcinoma as a location of the disease.
Patients and Methods
We retrospectively reviewed the charts of patients with a diagnosis of ophthalmic IgG4-RD, seen between 2009 and 2012 in the department of Ophthalmology and Internal Medicine of a single tertiary center. Clinical, biological, radiological, and histopathological features were reviewed. Patients presenting with typical manifestations of the disease, such as orbital or lacrimal gland infiltration, were excluded from the study. Diagnosis was based on histopathological analysis of tissues biopsies using hematoxylin–eosin staining, CD3, CD5, CD20, CD79, CD23, κ and λ light chain, and IgG and IgG4 immunostaining. Patients included had a positive IgG4 staining on histopathology, with IgG4+/IgG+ cell ratio >40%, and a negative biological and radiological workup for differentials such as sarcoidosis, lymphoma, and Wegener disease.
Results
Three patients presented with ophthalmic IgG4-RD, and 2 of them were included. The third patient presented with typical orbital involvement.
Case 1
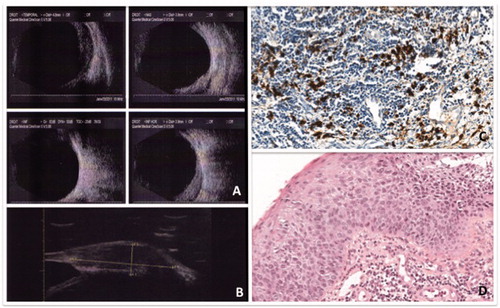
A 63-year-old Caucasian woman, with a history of thyroidectomy, presented with a 13-year history of recurrent unilateral nodular anterior and posterior scleritis, relapsing in spite of several courses of local topical corticosteroids and systemic nonsteroid anti-inflammatory treatment. Clinical examination showed normal visual acuity (20/20 OU), chemosis associated with lower corneal neovascularization, and normal fundus with no choroidal folds or serous retinal detachment. There were neither signs of lacrimal or orbital inflammation, intraocular inflammation, nor any systemic symptoms. Ocular B-scan ultrasonography and UBM confirmed the diagnostic of nodular anterior and posterior scleritis (). Systemic workup was negative for spondylarthritis, vasculitis, or granulomatosis. Lymphoma was excluded by a conjunctival biopsy showing nonspecific inflammatory infiltrate and normal medullar sample. Orbital MRI confirmed that the sclera was the only site of local inflammation. PET scan revealed small mediastinal lymphadenopathies. IgG4 serum rate was 135 mg/dL (normal <70 mg/dL) without hypergammaglobulinemia. A surgical scleral biopsy confirmed the diagnosis of IgG4-RD, demonstrating a lymphoplasmacytic infiltration with an IgG4+/IgG+ cell ratio of 50% (), but no signs of fibrosis or obliterative phlebitis. The patient was treated with high-dose corticosteroids (1 mg/kg/day of oral prednisone) allowing clinical improvement but relapsed after 2 months, requiring the adjunction of 20 mg/week of methotrexate to achieve satisfactory clinical outcome, a decrease of IgG4 serum rate to 46 mg/dL (normal <70 mg/dL), and tapering of the steroids over a period of 6 months (to 5 mg/d). Methotrexate dosage was slowly decreased over a year, and steroids were tapered to 3 mg/d without clinical or biological relapse.
FIGURE 1. Patient 1 presenting with chronic scleritis of the right eye: Panel A: B-ultrasonography of the right eye showing temporal and nasal thickening of the sclera. Panel B: UBM demonstrating nodular anterior nodular scleritis of the right eye. Panel C: Scleral biopsy of patient 1, x100 magnification. Lymphoplasmocytic B cells infiltration of the sclera. IgG4 immunostaining showing 50% of IgG4 + plasmocytes without any signs of fibrosis or vascularitis. Panel C: IgG4 staining showing important IgG4 deposit >50%. Panel D: Hematocylin eosin sections demonstrating massive lymphoplasmocytic infiltration.
Case 2
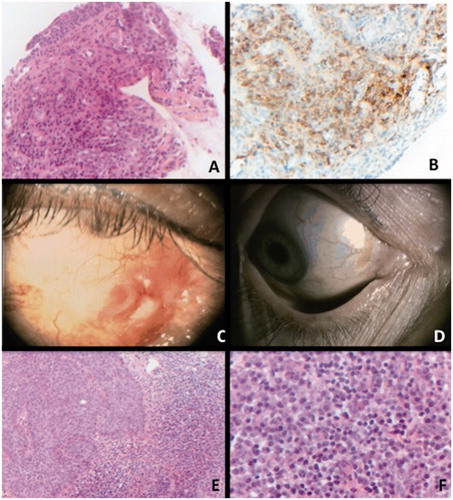
A 50-year-old Caucasian woman presented with a 2-year history of unilateral chronic conjunctival infiltration. Slit-lamp examination showed conjunctival discharge, chemosis, and conjunctival follicles, without corneal involvement. Visual acuity was 20/20 OU. There were no signs of intraocular inflammation on slit-lamp examination, neither orbital nor lacrimal infiltration. Several conjunctival smears were all negative for viral or bacterial infections. No signs of systemic involvement were clinically detected. Autoimmune and infectious workup were negative. IgG4 serum rate was 46 mg/dL (normal <70 mg/dL), and IgG2 (272 mg/dL) and IgG3 (43 mg/dL) levels were decreased. Conjunctival biopsy demonstrated lymphoplasmocytic infiltration with IgG4+/IgG+ cell ratio at 85% (). Neither storiform fibrosis nor obliterative phlebitis was observed. Full-body CT scan and PET scan found no signs of other organ involvement. Patient was started on a high dose of corticosteroids (1 mg/kg/d of prednisone), and inflammation and redness were initially controlled. After 1 month of follow-up, new lesions appeared on the site of the initial inflammation, presenting as red conjunctival nodules (). Conjunctival biopsy demonstrated the presence of an intraepithelial carcinoma (). Corticosteroids were slowly tapered over a period of 2 months and two cycles of topical 0.04% mitomycin C were administrated, allowing complete regression of the conjunctival carcinoma (). Due to local side effects of mitomycin with severe dry eye syndrome, the patient did not complete the third course of topical chemotherapy. No recurrence was observed after 2 years of follow-up.
FIGURE 2. Conjunctival biopsy of case 2. Panel A: Haematoxylin eosin staining, x200 magnification. Presence of a lymphoplasmocytic infiltration of the conjunctiva. Panel B: IgG4 immunostaining, x200 magnification.Diffuse staining is observed in the sample, composed with 85% of plasmocytes. Panel C: Clinical picture of patient 2 demonstrating conjunctival inflammation relapsing with in situ carcinoma after steroids treatment. Panel D: Complete regression of the carcinoma after 2 cycles of topical mytomicin. Panel E: Conjunctival biopsy of case 2 after apparent relapse of inflammation. Haematoxylin x100 left: epithelial dystrophy without crossing the basal membrane (in situ carcinoma), adjacent to a large lymphoplasmocytic infiltration. Panel F: x400 magnification. Plasmocytes with typical feature of cartwheel nucleus and light appearance of cytoplasm.
Discussion
IgG4-RD is a rare condition that is more frequent in Asia. Its prevalence is underestimated, mostly because of low awareness of the disease in western countries.Citation9 Mean age is typically within the fifth and sixth decade, as in our series,Citation9 and sex ratio (M/F) is 2.6/1.Citation3,Citation7 In our series, both patients were Caucasian women.
Orbital inflammation is a typical, well-described manifestation of IgG4-RD,Citation8,Citation9 often associated with lacrimal gland involvement.Citation10 Only 1 case of IgG4-RD involving the scleraCitation11 and 2 cases of conjunctival location of the disease have been reported: both conjunctival locations were associated with typical orbital inflammationCitation12 and eyelid lesions.Citation13 In our cases, atypical ophthalmic manifestations were the only manifestations of the disease. There were no signs of systemic or lacrymal involvement. In addition, this is the first case of conjunctival intraepithelial carcinoma occurring in ophthalmic IgG4-RD.
Histopathology of IgG4-RD is a crucial tool to establish the diagnosis,Citation14 showing massive CD4+ CD8+ polyclonal lymphoplasmacytic infiltration in lymphoid follicules,Citation3,Citation7 eosinophilic cells, and obliterative vasculitis.Citation9 In our 2 cases, histopathology found a typical lymphoplasmacytic IgG4+ infiltration but storiform fibrosis and phlebitis were absent. This can be explained by the smaller size of tissue samples compared to pancreatic samples. Scleral or conjunctival biopsies are not routinely performed in inflammatory diseases. Both patients in our study presented with a long-standing history of ophthalmological symptoms leading us to perform a biopsy with oriented immunostaining in order to achieve correct diagnosis and rule out alternative diagnosis.
Long diagnostic delay in IgG4-RD, between 3.8 years in the French cohortCitation9 and 7.5 years in our cases, is responsible for irreversible sequel due to extensive fibrosis.Citation15 Biopsies of the inflamed tissues are the only warrant of a prompt and accurate diagnosis in this condition. FDG-PET scan is the most useful radiological examination to investigate for other organs involvement and to find the most accessible sites to proceed to a biopsy. It also helps to eliminate alternative diagnosis or to estimate the inflammatory activity of the disease and its outcome during treatment.Citation16
Corticosteroids are the mainstay of treatment for IgG4-RD. Doses and duration of treatment are not consensual but suggested dosage is around 0.6 to 1 mg/kg/d with long tapering and maintenance dose between 2.5 and 10 mg/day of prednisone, in order to obtain initial remission, to lower relapse rateCitation7,Citation17 (50 to 25%), and to prevent short-term or long-term organ damage or further involvement of different organs.Citation18 There seems to be a limited window of time for corticosteroid efficiency.Citation17 In our series, diagnosis delay explains the initial response to corticosteroids but the early relapse despite the high dose of steroids administered. Moreover, patient 1 needed the use of an immunosuppressive drug to obtain good outcome. Treatment for relapsing and refractory cases is controversial.Citation7,Citation17 Methotrexate, azathioprine (2.0–2.5 mg/kg/d), mycophenolate mofetil (750 mg twice daily), cyclophosphamide, 6-mercaptopurine,Citation9 and bortezomib have been used in a few reports.Citation19 Since one of the hallmarks of the disease is a lymphoplasmocytic infiltration with CD20+ cells, anti-CD20 depletion with rituximab at doses of 1 g/15 days may be a treatment of choice, with a response rate of 90%Citation20 in refractory cases.
There are a few cases of malignancies arising in IgG4-RD sites reported in the literature. In some cases of ocular adnexal MALT lymphoma arising from IgG4-RD location,Citation21 histopathology showed that infiltration of IgG light chain restriction was surrounded by IgG4+ lymphoplasmacytic infiltrate without cellular atypies. Lymphoma is also considered as a possible complication of IgG4-related dacryoadenitis.Citation22 Association between chronic inflammation and non-Hodgkin lymphoma is well known, but it is difficult to differentiate between IgG4-related lymphoma and de novo lymphoma. It has been reported that 9% of 24 ocular adnexal marginal zone B-cell lymphomas were infiltrated by IgG4+ cells with significant sclerosis and serum abnormalities alike in systemic IgG4-RD.Citation23 Other malignancies have been reported at IgG4-RD sitesCitation4: cervical soft tissues marginal lymphoma, lung adenocarcinoma, parotidic adenocarcinoma,Citation24 urothelial in situ carcinoma, infiltrative ductular pancratic adenocarcinoma,Citation25 clear cell gastrointerstinal carcinoma, and T lymphoma developing 2 years after sclerosing cholangitis.Citation26 We report the first case of in situ conjunctival carcinoma arising in an IgG4 infiltrated site. Similarily to what has been reported in MALT-lymphoma arising from IgG4 location, we observed signs of chronic inflammation in the vicinity of the conjunctival carcinoma. This observation could lead to a diagnosis of pseudoepitheliomatous hyperplasia. However, the presence of atypical mitotic figures was in favor of a conjunctival carcinoma. Whether the carcinoma arose from an area of pseudoepitheliomatous hyperplasia is not known. Biopsies should be repeated if a new suspect lesion arises in a different location from the initial lesion during the course of the disease.
IgG4-RD is a new and rare entity, which can be challenging to diagnose, especially in case of atypical features such as conjunctival infiltration and scleritis. It should be suspected in the face of any chronic inflammatory ophthalmological manifestation, after excluding more frequent alternative diagnoses. A biopsy with appropriate staining can be mandatory to confirm the diagnostic and exclude a secondary malignancy.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738
- Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med. 2006;4:23
- Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2009;68:1310–1315
- Divatia M, Kim SA, Ro JY. IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J. 2012;53:15–34
- Sato Y, Notohara K, Kojima M, et al. IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int. 2010;60:247–258
- Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–3067 doi:10.1002/art.34593
- Masaki Y, Kurose N, Umehara H. IgG4-related disease: a novel lymphoproliferative disorder discovered and established in Japan in the 21st century. J Clin Exp Hematop JCEH. 2011;51:13–20
- Pasquali T, Schoenfield L, Spalding SJ, Singh AD. Orbital inflammation in IgG4-related sclerosing disease. Orbit Amst Neth. 2011;30:258–260
- Ebbo M, Daniel L, Pavic M, et al. IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine (Baltimore). 2012;91:49–56
- Sato Y, Ohshima K, Ichimura K, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008;58:465–470
- Ohno K, Sato Y, Ohshima K, et al. IgG4-related disease involving the sclera. Mod Rheumatol. 2012. doi:10.1007/s10165-012-0758-y
- Paulus YM, Cockerham KP, Cockerham GC, Gratzinger D. IgG4-positive sclerosing orbital inflammation involving the conjunctiva: a case report. Ocul Immunol Inflamm. 2012;20:375–377
- Da Fonseca FL, Ramos R de IP, de Lima PP, et al. Unilateral eyelid mass as an unusual presentation of ocular adnexal IgG4-related inflammation. Cornea. 2013;32:517–519
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol Off J United States Can Acad Pathol Inc. 2012;25:1181–1192
- Shimizu Y, Yamamoto M, Naishiro Y, et al. Necessity of early intervention for IgG4-related disease—delayed treatment induces fibrosis progression. Rheumatol Oxf Engl. 2013;52:679–683
- Nguyen VX, De Petris G, Nguyen BD. Usefulness of PET/CT imaging in systemic IgG4-related sclerosing disease: a report of three cases. JOP J Pancreas. 2011;12:297–305
- Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:67–71
- Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–1507
- Khan ML, Colby TV, Viggiano RW, Fonseca R. Treatment with bortezomib of a patient having hyper IgG4 disease. Clin Lymphoma Myeloma Leuk. 2010;10:217–219
- Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91:57–66
- Cheuk W, Yuen HKL, Chan ACL, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:1159–1167
- Cheuk W, Yuen HK, Chan JK. Complication of IgG4-related chronic sclerosing dacryoadenitis by lymphoma. Arch Ophthalmol. 2008 Aug;126(8):1170; author reply 1170. 2014 Jan;24(1):195--198
- Kubota T, Moritani S, Yoshino T, et al. Ocular adnexal marginal zone B cell lymphoma infiltrated by IgG4-positive plasma cells. J Clin Pathol. 2010;63:1059–1065
- Gill J, Angelo N, Yeong ML, McIvor N. Salivary duct carcinoma arising in IgG4-related autoimmune disease of the parotid gland. Hum Pathol. 2009;40:881–886doi:10.1016/j.humpath.2008.10.020
- Witkiewicz AK, Kennedy EP, Kennyon L, et al. Synchronous autoimmune pancreatitis and infiltrating pancreatic ductal adenocarcinoma: case report and review of the literature. Hum Pathol. 2008;39:1548–1551doi:10.1016/j.humpath.2008.01.021
- Kanda G, Ryu T, Shirai T, et al. Peripheral T-cell lymphoma that developed during the follow-up of IgG4-related disease. Intern Med Tokyo Jpn. 2011;50:155–160