To the Editor
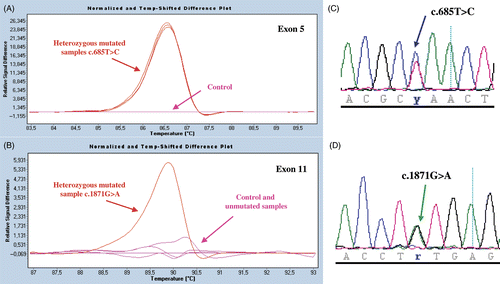
In 1999 we published the case of a male patient from the Bordeaux area of France with variant-type Glanzmann thrombasthenia (GT) linked to a heterozygous c.685T > C transition in exon 5 of ITGB3 giving rise to a L196P (L222P, including the leader sequence) substitution Citation[1], Citation[2]. This mutation was detected following an initial screening of the exons and splice sites of the ITGB3 and ITGA2B genes of the propositus (pat 1 here) by single-strand conformation polymorphism analysis of PCR products. All living close family members, namely his mother, sister and son were also heterozygous carriers of the mutation. While pat 1 showed the typical signs of GT with an absent aggregation to physiologic agonists, the other three family members all gave a normal response to ADP although retaining a delayed and poor response to collagen Citation[2]. This latter finding remains unexplained. Whereas his mother, son and sister have no bleeding symptoms, pat 1 has a lifelong history of mucocutaneous bleeding especially epistaxis. A sister died from nasal bleeding when she was a child. Whereas each of the three family members had somewhat decreased but >50% expression of αIIbβ3 in their platelets, pat 1 only possessed of the order of 8% of the normal platelet content of αIIbβ3 Citation[1]. Nevertheless, unlike for classic type I GT, his platelets contained detectable amounts of stored and presumably endocytosed Fg. An independent report on a second French family identified the presence of a homozygous ITGB3 c.685C > T nucleotide substitution and L196P substitution in a patient with about 10% of the expression of αIIbβ3 as revealed using a radiolabeled monoclonal antibody in binding studies Citation[3], Citation[4]. Again some α-granule stored Fg was present. All of the evidence therefore pointed to the presence of a second, undetected mutation in pat 1. Direct sequencing of all exons and splice sites of ITGA2B (30 exons) and ITGB3 (15 exons) by the French National Sequencing Center (Génoscope, Evry, France) while confirming the heterozygous ITGB3 exon 5 c.685C > T substitution for pat 1, has additionally shown the presence of a heterozygous ITGB3 exon 11 c.1871G > A transition leading to a C598Y (C624Y) substitution. A newly adapted high-resolution DNA melting curve (HRM) analysis procedure in the presence of a saturating DNA-binding dye Citation[5] and direct sequencing confirmed that whereas the heterozygous ITGB3 exon 5 substitution was indeed present in all of the family members, the propositus was the only family member to possess the exon 11 mutation ().
Figure 1. Mutation screening by HRM analysis and direct sequencing of PCR-amplified products. Results are shown for ITGB3 exons 5 and 11 for family members of pat 1. In panels A and B, we illustrate normalized and temperature-shifted melting curves of mutated and control PCR amplicons (Roche Light cycler 480 ResoLight Dye; Roche Diagnostics, Meylan, France) (c.685C > T, Leu196Pro; and c.1871G > A, Cys598Tyr). Control patterns (pink) mutated patterns (red) are clearly distinguished. Whereas the ITGB3 exon 5 substitution was present in all of the family members (see superimposed lines), the propositus was the only family member to possess the exon 11 mutation. In panels C and D are shown the sequencing profiles of respective heterozygous mutated PCR products. Methodological details will be supplied on request.
A heterozygous ITGB3 c.1871G > A transition leading to a C598Y substitution has been previously reported for a third unrelated French patient (pat 2 of this study), where it was found together with an ITGB3 exon 5 c.724C > T nonsense mutation giving an R216X (R242X) stop codon Citation[6]. Historically, this patient was one of the first GT patients to be characterized and possessed of the order of 10% residual αIIbβ3 able to bind fibrinogen (Fg) when platelets were incubated with ADP Citation[7–11]. The effects of the L196P and C598Y substitutions on β3 function have both been previously examined by site-directed mutagenesis followed by transfection in CHO cells Citation[4], Citation[12]. The presence of 196P within the I-domain of β3 is inhibitory, and blocks the Fg-binding capacity of both αIIbβ3 and αvβ3. In fact, this is one of the few mutations where a dual effect on both β3 integrins has been clearly established. In contrast, the C598Y mutation, like many of the β3 cysteine mutations in the cysteine-rich EGF domains (C598 forms a disulfide with C588 in EGF-4) is partially activating, allowing the spontaneous binding of the activation-dependent monoclonal antibody PAC-1 to transfected CHO or BHK cells but not the binding of soluble Fg Citation[12], Citation[13]. It should be noted that whereas the residual αIIbβ3 of pat 1 would be expected to be a mixture of the integrin containing either β3P196 or β3Y598, that of patient 2 is likely to contain only β3Y598 (the second mutation is a stop codon giving rise to a severely truncated β3 not present in her platelets). Thus while both inhibitory and partially activating β3 subunits prevent normal αIIbβ3 maturation in megakaryocytes, possibly by delaying transport through the Golgi apparatus Citation[14], residual integrin surface expression does occur. Of note is the fact that platelets of each patient contain substantial amounts of α-granule stored Fg, implying that a functional β3 I- domain or intact cysteine-rich EGF domains are not required for Fg uptake. It is highly probable that the inability of platelets of pat 2 to aggregate is due to the low density of αIIbβ3 on the platelet surface. Also of interest is that patient 2 has not been clinically protected by the presence of residual partially active integrin as she has a life-long history of mucocutaneous bleeding intermingled with several severe episodes requiring transfusions (pat 41 in Citation[15]). Whether or not the C598Y mutation affects αvβ3 expression or function is not known.
In conclusion, our study helps define the pathophysiology of two rare ITGB3 mutations giving rise to GT, and describe the first patient with a qualitative αIIbβ3 deficiency with both inhibitory and partially activating amino acid substitutions on the β3 subunit.
Acknowledgements
This study was financed by contract N° AP07/08.42 with the Génoscope d'Evry and from INSERM (ANR-08-GENO-028-03). Informed consent was obtained and the study performed under the national ethical guidelines of the CRPP.
References
- Nurden AT, Ruan J, Pasquet J-M, Gauthier B, Combrié R, Kunicki T, Nurden P. A novel 196Leu to Pro substitution in the b3 subunit of the αIIbβ3 integrin in a patient with a variant form of Glanzmann thrombasthenia. Platelets 2002; 13: 101–111
- Nurden A, Jacquelin B, Tuleja E, Combrie R, Nurden P. Reduced collagen-induced platelet aggregation in obligate heterozygotes of a Glanzmann thrombasthenia variant with a β3 mutation. Thromb Haemost 2002; 88: 364–365
- Morel-Kopp M-C, Lecompte T, Schlegel N, Hivert P, Kaplan C. Use of murine monoclonal antibodies to study thrombopathies related to GP IIb/IIIa complexes. Platelet immunology: clinical and fundamental aspects, C Kaplan-Gouet, N Schlegel, C Salmon, J McGregor. John Libbey, Paris 1991; 161–171
- Morel-Kopp M-C, Melchior C, Chen P, Ammerlaan W, Lecompte T, Kaplan C, Kieffer N. A naturally occurring point mutation in the β3 integrin MIDAS-like domain affects αvβ3 and αIIbβ3 receptor function. Thromb Haemost 2001; 86: 1425–1434
- Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 1997; 245: 154–160
- Schlegel N, Chen P, Binard S, Maisonneuve P, Rosa JP, Kieffer N, Caen JP. Type II Glanzmann thrombasthenia (GT) in a compound heterozygote for the glycoprotein (GP) IIIa gene associating two novel mutations, a stop codon (Arg216) and a Cys598Tyr substitution in mature GPIIIa. Blood 1999; 94(Suppl 1)451a
- Caen JP, Castaldi PA, Leclerc JC, Inceman S, Larrieu MJ, Probst M, Bernard J. Congenital bleeding disorders with long bleeding time and normal platelet count. I. Glanzmann's thrombasthenia. Am J Med 1966; 41: 4–26
- Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in three cases of Glanzmann's thrombasthenia. Br J Haematol 1974; 28: 253–260
- Hagen I, Nurden AT, Bjerrum OJ, Solum NO, Caen JP. Immunochemical evidence for protein abnormalities from patients with Glanzmann's thrombasthenia and Bernard-Soulier syndrome. J Clin Invest 1980; 65: 22–29
- Lee H, Nurden AT, Thomaidis A, Caen JP. Relationship between fibrinogen binding and the platelet glycoprotein deficiencies in Glanzmann thrombasthenia type I and type II. Br J Haematol 1981; 48: 47–57
- Nurden AT, Didry D, Kieffer N, McEver RP. Residual amounts of glycoproteins IIb and IIIa may be present in the platelets of most patients with Glanzmann's thrombasthenia. Blood 1985; 65: 1021–1024
- Chen P, Melchior C, Brons NHC, Schlegel N, Caen JC, Kieffer N. Probing conformational changes in the I-like domain and the cysteine-rich repeat of human β3 integrins following disulfide bond disruption by cysteine mutations. Identification of cysteine 598 involved in αIIbβ3 activation. J Biol Chem 2001; 276: 42: 38628–38635
- Mor-Cohen R, Rosenberg N, Landau N, Lahav J, Seligsohn U. Specific cysteines in β3 are involved in disulfide bond exhange-dependent and -independent activation of αIIbβ3. J Biol Chem 2008; 283: 19235–19244
- Mitchell WB, Li J, Murcia M, Valentin N, Newman PJ, Coller BS. Mapping early conformational changes in alphaIIb and beta3 during biogenesis reveals a potential mechanism for alphaIIbbeta3 adopting its bent conformation. Blood 2007; 109: 3725–3732
- George JN, Caen JP, Nurden AT. Glanzmann's thrombasthenia: The spectrum of clinical disease. Blood 1990; 75: 1383–1395