
To the editor,
We studied two related patients (III.4 and III.10) with spontaneous clinical bleeding problems and a family history suggestive of X-linked inherited thrombocytopenia (). Both patients presented with hematomas, ecchymosis, epistaxis, and gingival bleeding symptoms since childhood and developed splenomegaly. Hematological investigations (after the patient was splenectomized) showed the presence of macrothrombocytopenia combined with mild features of dyserythropoiesis including poikilocytosis, anisocytosis, and schizocytes, Howell–Jolly bodies in erythrocytes and stomatocytes but no anemia (). Both patients did not respond to immune thrombocytopenic purpura-specific therapy such as intravenous immunoglobulins, steroids, or splenectomy. Bone marrow analysis showed hypermegakaryopoiesis with an increased number of immature megakaryoblasts but no features of dysmyelopoiesis. Platelet functional studies for patient III:4 showed normal aggregations for collagen and ristocetin (amplitudes of 84% and 86%, respectively) and mildly reduced for ADP (amplitude of 40%). The PFA100 closure time with collagen/epinephrine for patient III:4 was prolonged (>300 s; nl: 82–142 s) while normal with collagen/ADP.
Figure 1. (A) Patients with thrombocytopenia are represented by filled symbols. (B) Schematic presentation of the N-terminal zinc finger of GATA1 with the mutations that are reported and the D218N mutation presented in the present study. (C) Sequencing analysis of GATA1 exon 4 using gDNA of patient III:4 and a control male. A basepair substitution G to A was found that converted aspartic acid (D) into asparagine (N). (D) Platelet count profiles for patients with the different D218 GATA1 mutations and an unrelated healthy control subject. The histogram plots present the platelet count (PLT) in function of the mean platelet volume (MPV) as indicated.
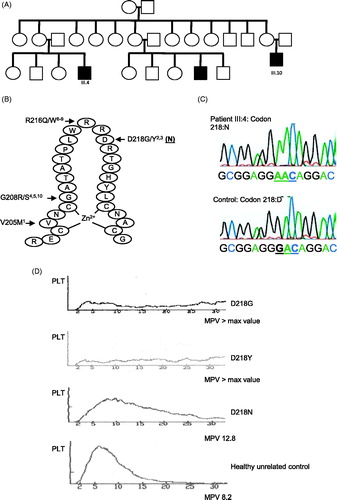
Table I. Hematological profiles of patients with GATA D218 mutations.
X-linked macrothrombocytopenia with platelet dysfunction has been described by others and us for patients with mutations in the gene for the transcription factor GATA1 [Citation1–10]. The GATA family of nuclear regulatory proteins serves as a prototype for the action of lineage-restricted transcription factors. The GATA1 protein is typically present in the more differentiated cells of the erythroid, megakaryocytic, mast, and eosinophilic lineages, whereas GATA2 expression is limited to HSCs and early haematopoietic progenitors [Citation11]. Remarkably since its first description in 2001, still only a limited number of families are presently reported with germline GATA1 missense mutations and these presented with closely related but still somewhat altered hematological disorders. They comprise severe dyserythropoietic anemia with macrothrombocytopenia (V205M, G208R, and D218Y), macrothrombocytopenia with mild dyserythropoietic features (G208S, D218G), macrothrombocytopenia with mild β-thalassemia (R216Q), and congenital erythropoietic porphyria (R216W) [Citation1–10]. shows the position of these mutations that are all present in the N-terminal zinc finger of GATA1. Males with these mutations have enlarged platelets with a paucity of alpha granules and a reduced function. Some patients also present with splenomegaly [Citation7, Citation9] or myelodysplasia with marked organomegaly [Citation10], likely due to ineffective and consequential extramedullary hematopoiesis. Genotype–phenotype correlations are hampered by the low numbers of families with a GATA1 mutation to date, with only two mutations (R216Q [Citation6–8] and G208R [Citation5, Citation10]) being identified in more than one family.
Sanger sequencing of the different GATA1 exons using leukocyte gDNA of patient III:4 showed the presence of a missense mutation gac (D) to aac (N) at codon 218 (). Interestingly, we already have described two other missense mutations in this residue for two unrelated families for which patients presented with mild macrothrombocytopenia with dyserythropoiesis (D218G) [Citation2] and severe macrothrombocytopenia with anemia (D218Y) [Citation3]. A genomic DNA sample was not available from subject III:10 to confirm the presence to the GATA1 D218N variant. The D218N GATA1 variant was not present in the dbSNP or 1000 genomes databases (www.1000genomes.org). In addition, SIFT (sift.jcvi.org) predicted GATA1 variants D218G/Y and N to be damaging.
The platelet defect for propositus III:4 and his relative III:10 seems milder than found for subjects with the D218G or D218Y mutations as the degree of thrombocytopenia is less severe and especially their platelets are large but not giant ( and ). Actually, without having evidence of an X-linked inheritance patterns in this family, a single patient with this type of platelet defect might escape the genetic diagnosis of GATA1. This could be a reason why still only few patients have been described with GATA1 mutations still giving us no idea about the frequency of this disorder. Therefore, it remains important to report also these milder cases.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was supported by research grants G.0490.10 N, G.0743.09, and G.0B17.13 N from the Fund for Scientific Research – Flanders (FWO-Vlaanderen, Belgium) and GOA/2009/13 from the Research Council of the University of Leuven (Onderzoeksraad KULeuven‚ Belgium). CVG is holder of a clinical-fundamental research mandate of the FWO-Vlaanderen, Belgium and of the Bayer and Norbert Heimburger (CSL Behring) Chairs.
References
- Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, Weiss MJ. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet 2000;24:266–270
- Freson K, Devriendt K, Matthijs G, Van Hoof A, De Vos R, Thys C, Minner K, Hoylaerts MF, Vermylen J, Van Geet C. Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood 2001;98:85–92
- Freson K, Matthijs G, Thys C, Marien P, Hoylaerts MF, Vermylen J, Van Geet C. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum Mol Genet 2002;11:147–152
- Mehaffey MG, Newton AL, Gandhi MJ, Crossley M, Drachman JG. X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood 2001;98:2681–2688
- Del Vecchio GC, Giordani L, De Santis A, De Mattia D. Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol 2005;114:113–116
- Yu C, Niakan KK, Matsushita M, Stamatoyannopoulos G, Orkin SH, Raskind WH. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood 2002;100:2040–2045
- Balduini CL, Pecci A, Loffredo G, Izzo P, Noris P, Grosso M, Bergamaschi G, Rosti V, Magrini U, Ceresa IF, et al. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb Haemost 2004;91:129–140
- Tubman VN, Levine JE, Campagna DR, Monahan-Earley R, Dvorak AM, Neufeld EJ, Fleming MD. X-linked gray platelet syndrome due to a GATA1 Arg216Gln mutation. Blood 2007;109:3297–3299
- Phillips JD, Steensma DP, Pulsipher MA, Spangrude GJ, Kushner JP. Congenital erythropoietic porphyria due to a mutation in GATA1: The first trans-acting mutation causative for a human porphyria. Blood 2007;109:2618–2621
- Kratz CP, Niemeyer CM, Karow A, Volz-Fleckenstein M, Schmitt-Gräff A, Strahm B. Congenital transfusion-dependent anemia and thrombocytopenia with myelodysplasia due to a recurrent GATA1(G208R) germline mutation. Leukemia 2008;22:432–434
- Bresnick EH, Martowicz ML, Pal S, Johnson KD. Developmental control via GATA factor interplay at chromatin domains. J Cell Physiol 2005;205:1–9