Abstract
The hERG potassium channel is a member of the voltage gated potassium (Kv) channel family, comprising a pore domain and four voltage sensing domains (VSDs). Like other Kv channels, the VSD senses changes in membrane voltage and transmits the signal to gates located in the pore domain; the gates open at positive potentials (activation) and close at negative potentials, thereby controlling the ion flux. hERG, however, differs from other Kv channels in that it is activated slowly but inactivated rapidly – a property that is crucial for the role it plays in the repolarization of the cardiac action potential. Voltage-gating requires movement of gating charges across the membrane electric field, which is accomplished by the transmembrane movement of the fourth transmembrane segment, S4, of the VSD containing the positively charged arginine or lysine residues. Here we ask if the functional differences between hERG and other Kv channels could arise from differences in the transmembrane movement of S4. To address this, we have introduced single cysteine residues into the S4 region of the VSD, expressed the mutant channels in Xenopus oocytes and examined the effect of membrane impermeable para-chloromercuribenzene sulphonate on function by the two-electrode voltage clamp technique. Our results show that depolarization results in the accessibility of seven consecutive S4 residues, including the first two charged residues, K525 and R528, to extracellularly applied reagent. These data indicate that the extent of S4 movement in hERG is similar to other Kv channels, including the archabacterial KvAP and the Shaker channel of Drosophila.
Introduction
The human ether-á-go-go related gene (hERG) channel, encoded by the KCNH2 gene, plays a key role in the repolarization of the cardiac action potential [Citation1–4]. hERG belongs to the voltage-gated potassium channel (Kv) family, and like other Kv channels, it is activated by membrane depolarization. However, hERG differs from other Kv channels in its gating properties: During depolarization, it is activated very slowly, but undergoes rapid (C-type) inactivation. Upon repolarization it recovers from inactivation very rapidly, but deactivates slowly, allowing K+ efflux [Citation5–7]. These unique kinetics result in inward rectification, a property that limits K+ efflux during the plateau phase, but allows K+ flux during the repolarization phase of the action potential. Furthermore, inactivation of hERG is intrinsically voltage sensitive and is not coupled to activation in the same way as in Kv channels [Citation5–7]. The importance of these unique biophysical properties to the regulation of cardiac rhythm is reflected by the fact that genetic mutations (http://www.fsm.it/cardmoc/) that alter the properties of hERG lead to the Long QT syndrome 2 (OMIM:152427), a condition that predisposes the affected individuals to ventricular arrhythmias and sudden death [Citation2–4]. The structural features and conformational changes that contribute to the differences in biophysical properties between hERG and other Kv channels are not fully understood. Major advances have recently been made in our understanding of the structure of Kv channels as well as how they respond to changes in membrane voltage [Citation8–10]. All Kv channels are tetramers, each subunit comprising six transmembrane segments, S1–S6, with intracellular N- and C-terminal domains. S5–S6 from the four subunits forms the central ion conducting pore domain. The pore is surrounded by four voltage sensing domains (VSD), made from S1–S4 [Citation11]. S4 contains 6–7 positively charged residues (arginine/lysine). The crystal structures of rat Kv1.2 (presumed open state) [Citation11], KvAP, an archaebacterial Kv channel [Citation12], and the isolated VSD of KvAP [Citation12] have been reported. Emerging evidence suggests that the helix-loop-helix structure comprising S3b (C-terminal end of S3), S4 and the connecting loop, termed the ‘paddle’ is conserved among all Kv channels and is the key component of the voltage sensing machinery of all voltage-gated ion channels [Citation13–15].
It is now well established that in response to membrane depolarization S4 moves outwards taking several charged residues (gating charges) out of the membrane electric field (bilayer) [Citation16–18]. This movement is coupled to the opening of channel gates located at the intracellular aspect of the channel via the S4–S5 linker, leading to K+ flux. While the movement of S4 is well documented, it is hotly debated as to whether it moves as an independent domain or as an rigid unit with S3b, that is, as a paddle [Citation10,Citation19,Citation20].
Studies have examined the movement of S4 in hERG [Citation21–23] in an attempt to explain the differences in the gating properties between hERG and the conventional Kv channels. Using voltage-clamp fluorescence measurements, Smith and Yellen reported that the outward movement of S4 in hERG is slower than that in the Shaker Kv channel; the authors suggested that this may account for the slow activation of hERG [Citation22]. This conclusion is consistent with the subsequent report that the rise of gating current during activation of hERG is approximately two orders of magnitude slower than that for the Shaker Kv channel [Citation23]. The molecular basis for the slow movement of S4 in hERG is not entirely clear, but unlike Kv channels, which have three conserved negative charges, hERG has six negative charges in the VSD, including the three at the conserved positions (see ). In Kv channels the three conserved negative charges are thought to interact differentially with the positive charges of S4 in the closed and open states of the channel and thereby play a role in S4 motion. It seems plausible that the additional negative charges in the VSD could slow down the motion of S4 in hERG by forming further salt bridges [Citation24].
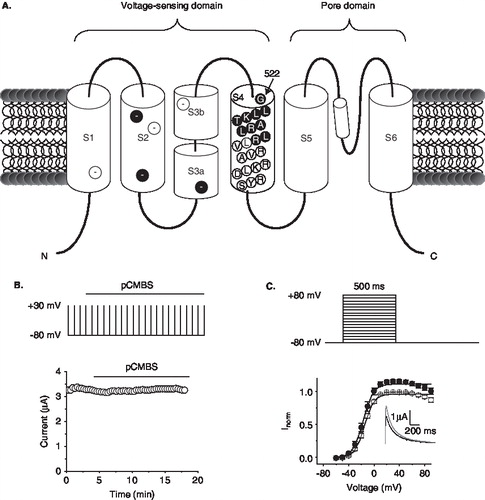
Figure 1. Effect of pCMBS on wild-type hERG channels. (A) Schematic of the topology of a single hERG subunit. Approximate location of negative charges in S1–S3 is shown in filled circles for conserved positions, and in open circles for positions unique to hERG; residues of S4 tested in this study (522–531) are shown in filled circles in S4. (B) Time course of the effect of pCMBS on wild-type hERG. Top, schematic of the repeated pulse protocol used to measure hERG currents. Cells were depolarized to +30 mV for 500 ms from −80 mV at intervals of 20 s. Peak tail currents were measured at −80 mV following each depolarizing step. Horizontal bar indicates time period over which 100 µM pCMBS was applied. Bottom graph shows representative data for wild-type hERG obtained using the pulse protocol. (C) Top, Schematic of the I-V protocol; cells were depolarized for 500 ms to voltages between −60 mV and 80 mV in 10 mV steps from a h.p. of −80 mV; steps were separated by 20 s. Bottom, normalized I-V relationships for wild-type channels calculated from peak currents recorded at −80 mV following each depolarizing step, before (○) and after (•) exposure to 100 µM pCMBS. The smooth curves correspond to data fitted to the Boltzmann function; n = 3. Inset shows current traces recorded at −80 mV following an 80 mV step, before (black trace) and after (grey trace) after exposure to 100 µM pCMBS.
Another difference between the Shaker Kv channel and hERG is the amount of gating charge (eo) translocated across the membrane electric field: in Shaker it is 12–13 eo, whereas in hERG the value is 8 eo [Citation21]. This suggests that either the membrane embedded portion of S4 in the resting state of hERG has fewer positive charges, or the extent of S4 motion (hence the number of charges that move out of the membrane) is smaller than that in the Shaker. But the predicted sequence of S4 in hERG has the same number of charges as in Shaker. However, there is no information on how many of these charges lie within the membrane in the resting state of the channel, or on how far S4 moves during depolarization.
Here, we set out to address these questions by examining the accessibility of the N-terminal portion of S4 to extracellular solvent at the resting and depolarizing potentials. The results show that the extent of exposure of S4 in hERG during depolarization is similar to that of the Shaker Kv channel and KvAP, which includes the first two charged residues.
Materials and methods
Mutagenesis and expression
Mutations were introduced into the hERG cDNA clone (Acc. No. GI:4557729) in the pSP64 vector by the QuikChange method (Stratagene). cRNA was prepared from hERG and its mutants using the SP6 Megascript kit (Ambion) and injected into the stage V or VI oocytes isolated from Xenopus toads, killed by cervical dislocation after anaesthetization with 3-amino-benzoic acid ethyl ester [Citation17]. Oocytes were incubated in ND-96 solution (in mM: NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5, sodium pyruvate 2.5, pH 7.5) supplemented with 100 µM DTT solution for 1–3 days at 18°C before recording currents. The details of the methods are as described [Citation17].
Electrophysiology
Whole cell currents were measured using a two-electrode voltage clamp (TEVC) in Ringer’s solution containing (mM): 115 NaCl, 2 KCl, 1.8 CaCl2, 10 HEPES, pH 7.2. Microelectrodes were made from borosilicate glass, filled with 3 M KCl, and had resistances between 0.5 and 2.0 MΩ. The details of the methods are as described [Citation17]. Current (I)-voltage (V) relationships were measured from oocytes using the standard tail current analysis. Oocytes were depolarized to test potentials in the range −80 mV to +80 mV in 10 mV steps from a holding potential of −80 mV; the steps were applied at 20 s intervals and lasted for 500 ms. Tail currents were recorded at −80 mV. Currents were normalized to the maximum current value (Imax) and fitted with a Boltzmann function: I/Imax = [1 + exp((V0.5-V)/k)] − 1, where I/Imax is the relative tail current, V is the test potential, V0.5 is the midpoint of half maximal current and k is the slope factor (= RT/zF, where R is the gas constant, T the absolute temperature, z the valence, and F the Faraday constant).
After measuring I-V relationships in Ringers, effect of para-chloromercuribenzene sulphonate (pCMBS, made up in Ringers solution) on channel tail currents was measured using a repeated pulse protocol. For this, control currents were first measured in Ringer’s solution during repeated depolarizing steps (+30 mV) given from a holding potential of -80 mV. The cells were then superfused with pCMBS (100 μM) and current recordings were continued until a steady-state effect was achieved. Following this, I-V relationships were again recorded.
To investigate the voltage dependence of the pCMBS effects, after measuring control tail currents during the depolarizing steps (as mentioned above), cells were held at −80 mV for 30 s to allow the channels to recover from inactivation and attain the resting state. pCMBS was then applied while holding at this potential for 10 min before recommencing current measurements, in the continued presence of pCMBS until a steady-state effect was achieved. All experiments were repeated at least three times (n ≥ 3), and the data presented are either representative or mean ± SEM. Significance (p) of any effects was calculated using a one tailed Student’s t-test. A p value of < 0.05 was considered significant.
Results
Accessibility of hERG S4 cysteines to extracellularly applied pCMBS
In order to identify which S4 positions are accessible to extracellular solvent, we have used the substituted cysteine accessibility method (SCAM) [Citation16,Citation17]. For this, we have engineered single cysteines into several consecutive positions (522–531) in the outer half of the S4 segment of hERG (). We then expressed each mutant channel in Xenopus occytes and examined the effect of pCMBS on currents using two-electrode voltage clamp. The rationale of the approach was that being membrane impermeable, pCMBS will react with cysteines only when they are located in the extracellular phase [Citation17]. We followed the reaction by measuring inhibition of currents during the application of the reagent. If an inhibition was observed, we interpreted that the position occupied by the engineered cysteine is accessible to extracellular solvent.
and C show the effect of application of pCMBS (100 µM) on currents through the wild-type hERG channel. No effect was seen on currents elicited during repeated step depolarizing pulses (), but a slight increase in current was observed at positive potentials in I–V curves (). This result is consistent with the absence of cysteines in the extracellular portion of the channel. shows the results for mutant channels. All mutant channels tested gave rise to robust currents except K525C; further studies were not attempted on K525C because of low currents. Cysteine substitution caused changes in the voltage dependence of channel activation for several positions. For some, V1/2, the voltage for half maximal activation, was shifted in the negative direction relative to the wild-type hERG (G522C, −7.7 ± 2.8; L523C, −6.4 ± 1.8; T526C, −6.4 ± 2.4); for others the shift was positive (L524C, 20.9 ± ï 3.8; R528, 33.5 ± ï 2.9; L529C, 12.1 ± 2.5; L530, 21.8 ± 1.4; R531, 50 ± ï 2.1). As expected, the cysteine mutants of charged residues showed a positive shift in the voltage dependence of channel activation (see Supplementary Figure S1, online version only, for a comparison with the Shaker channel).
Figure 2. Accessibility of substituted S4 cysteines to extracellular pCMBS. Effect of pCMBS on the I-V relationships of indicated S4 hERG cysteine mutant channels measured as described in the legend to . Inset shows current traces recorded at −80 mV following an 80 mV step, before (black trace) and after (grey trace) after exposure to 100 µM pCMBS. pCMBS inhibited currents through G522C to L529C channels significantly (p < 0.05), but has no effect on L530C and R531C (p ≥ 0.05).
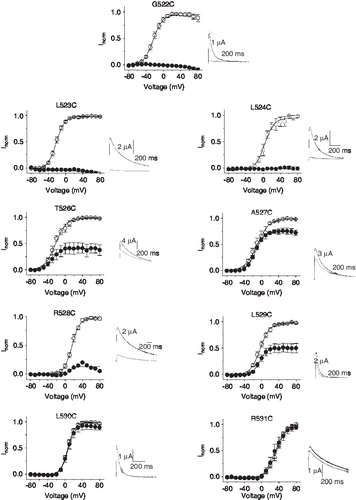
In contrast to the wild-type channel, channels containing engineered cysteines at positions 522–529 (with the exception of position 525), were all significantly (p ≤ 0.05) inhibited by pCMBS, although the degree of inhibition varied between different mutants (22.5 ± 4.5% for 527C to almost complete inhibition for 522C, 523C and 524C). Mutants beyond position 529, namely, 530C and 531C, were unaffected by pCMBS (). These data indicate that positions 522–529 are accessible to extracellular solvent at some point during gating, but do not suggest the state in which each of these residues access the solvent because during the application of pCMBS, the channel would be fluctuating between the resting state (−80 mV) and the activated/inactivated state (positive potential). Although we do not have data for 525C, we would expect this residue to be exposed because several (3–4) residues on either side of this position are accessible to extracellular solvent. Furthermore, Zhang et al, who were able to measure currents for the 525C mutant channel, showed that MTSET slows the deactivation kinetics of the channel [Citation21].
State-dependence of accessibility of S4 cysteines
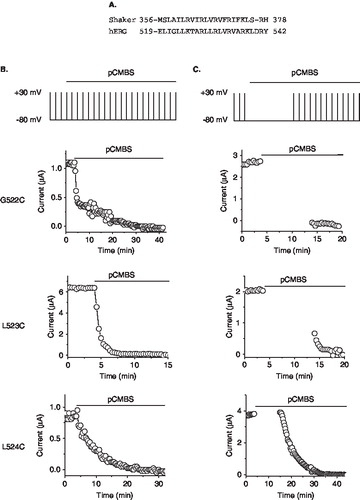
Previous studies with other Kv channels have shown that a number of residues at the top of S4 remain buried in the membrane in the resting state, but move into the extracellular solvent during depolarization [Citation16,Citation17,Citation19,Citation25]. In the Shaker channel, cysteine replacing A359 of the S4 helix is accessible to extracellular pCMBS in the resting state; by contrast, cysteines introduced at positions 361 (L) and beyond are not accessible in the resting state, but become accessible upon activation [Citation17,Citation20]. Sequence alignment shows that residues A359 and L361 of Shaker correspond to G522 and L524 of hERG (). We therefore examined the state-dependent accessibility of cysteines at positions 522 and 524, along with the middle 523 position. For this, we first estimated the time required for complete inhibition of each mutant by 100 µM pCMBS, applied during repeated test potentials elicited from the holding potential of −80 mV (). Using separate oocytes, we then applied the reagent while holding the oocyte at −80 mV for a period long enough to produce almost complete inhibition before commencing repeated test pulses. Results presented in show that pCMBS inhibits the 522C mutant channel completely at −80 mV indicating that this residue is solvent exposed in the resting state of the channel. By contrast, the 524C mutant channel was unaffected by pCMBS at −80 mV, but was completely inhibited during the subsequent depolarizing pulses. This indicates that position 524 is buried in the membrane in the resting state of the channel, but is exposed to extracellular phase during depolarization. 523C is partially inhibited during pCMBS application at −80 mV, which could suggest that this residue is likely near the interface between the membrane and the extracellular solvent.
Figure 3. State-dependent accessibility of S4 cysteines to extracellular pCMBS. (A) Sequence alignment of the S4 region of the Shaker (GI: 24642914) and hERG (GI: 4557729) potassium channels. (B) Time course of the effect of pCMBS on currents through the indicated hERG S4 cysteine mutant channels, measured as described in the legend to Figure 1B. (C) State-dependence of pCMBS effect. Top, protocol used to determine the state-dependence: Current recordings were made as in Figure 1B except that after recording control currents, cells where held at −80 mV with no depolarizing steps for 10 min (to keep the channels in the closed state; see Methods), before resuming depolarizing pulses. pCMBS application began whilst cells were at held at −80 mV as indicated by the horizontal bar. pCMBS reacted G522C, L523C, but not L524 at −80 mV, as indicated by a change in current amplitude measured immediately after the −80 mV holding step (gap in data points).
Discussion
The hERG potassium channel belongs to the family of Kv channels, but it displays a number of differences from other Kv channels [Citation3,Citation4]. First, hERG exhibits slow activation and deactivation compared with rapid inactivation and recovery from inactivation [Citation5,Citation6]. Second, inactivation of hERG is intrinsically voltage sensitive and is not coupled to activation in the same way as in other Kv channels. Third, during gating, fewer gating charges move across the membrane electric field when compared with the Shaker channel [Citation21]. Fourth, voltage-clamp fluorimetry studies [Citation22] and gating current kinetics [Citation23] suggested that S4 moves more slowly in hERG than in Kv channels. These differences in biophysical properties, together with the unique structural differences between hERG and other Kv channels (especially the presence of excess negative charges in the VSD) raise the key question: are the voltage-induced conformational changes in hERG different from other Kv channels?
To address this, we examined the transmembrane movement of S4 using the cysteine accessibility approach on hERG. Our data show that at negative potentials, when the channel is in its resting state, the extracellular membrane boundary of S4 lies at position 523 as this is the point at which substituted cysteine becomes dependent on depolarization for the membrane impermeable cysteine modification reagent pCMBS to react (). During depolarization, when the channel undergoes slow activation, but fast inactivation, the extracellular membrane boundary changes to be between positions 529 and 530, as substituted cysteine residues at position 530 and below appear to be unable to access pCMBS (). Thus during depolarization, residues 523–529 of S4 move out of the membrane bilayer into the extracellular solvent phase. Although we cannot suggest how much of this movement is associated with inactivation, studies with Kv channels suggest that the majority of S4 exposure occurs during activation and that changes in S4 exposure during C-type inactivation are minimal [Citation8,Citation10,Citation26].
The stretch of accessible hERG S4 residues includes two charged positions, K525 and R528. This result is in disagreement with a previous accessibility study, which reported that cysteine at position 525, but not at 528, becomes accessible to MTSET during depolarization [Citation21]. In view of these differences, we have tested the effect of MTSET on R528C, along with two other mutant channels (G522C and L530C). However, none of the mutants appear to be affected by the MTSET reagent (Supplementary Figure S2, online version only). Furthermore, application of pCMBS at a depolarizing potential (+20 mV), using the protocol described by Zhang et al. [Citation21], resulted in almost complete inhibition of currents through the R528C mutant channel; by contrast, MTSET showed no effect on this mutant under these conditions (Supplementary Figure S3, online version only), a result that is in agreement with the findings of Zhang et al. The reason for the difference between the two results is unclear, but could lie in the nature of the reagent used. It is possible that MTSET might have reacted with G522C and R528C, but had no detectable effect on the biophysical properties of the channel. A previous study with the Shaker channel indeed reported that MTSET could react with substituted cysteines but produce no effect on the function of the channel [Citation27].
Based on the lack of effect of MTSET on the R528C hERG mutant channel (see above), Zhang et al. proposed that in hERG charge is absent at the first position of S4 and that K525 corresponds to the second charged position of Shaker, which is R365 [Citation21]. Accordingly, their alignment places I360 of Shaker in line with L520 of hERG. The authors concluded that the absence of charge at the first position could account for the ∼ 4–5 eo difference in the gating charge translocated across the membrane electric field between Shaker (12–13 eo) and hERG (8 eo) [Citation21]. However, when we aligned all the accessible residues of hERG against Shaker, the first two charged residues in Shaker, R362 and R365, correspond to K525 and R528 of hERG respectively (). Moreover, I360 in Shaker aligns with L523 in hERG, which is in perfect agreement with the evidence that I360 in Shaker (27) and L523 in hERG () occur near the outer membrane-aqueous interface. We therefore conclude that charge is present at the first position (525) of membrane-embedded portion of hERG S4, and since it moves out the membrane bilayer, we expect that K525 would contribute to the gating charge in the same way as in Shaker. Thus the origin of the difference in the eo values between Shaker and hERG remains to be established. Structural differences in other parts of the voltage sensor, including the additional negative charges, or configuration of water crevices surrounding the cytoplasmic end of the sensor [Citation16,Citation28] might contribute to the differences in eo values between hERG and Shaker. One intriguing possibility is that the aqueous crevice at the intracellular phase of S4 could be smaller in hERG compared with other Kv channels, as this would increase the size of the electric field across which S4 charges need to move, and thereby reduce the net gating charge translocated.
We next compare S4 motion in hERG with studies in other Kv channels (). The majority of studies on voltage sensing were performed on S4 of the Shaker Kv channel [Citation3,Citation8–10,Citation16,Citation17,Citation25,Citation27] and more recently on S4 of the archaebacterial KvAP [Citation19,Citation26]. Interestingly, the extent of S4 exposure in hERG turned out to be remarkably similar to that reported in the Shaker channel, when pCMBS was used as a probe [Citation17,Citation27]. In both the channels seven positions become accessible to pCMBS during membrane depolarization. In KvAP, where capture of biotin substituted at different positions of S4 by avidin was examined, remarkably, the same number of positions moves into the extracellular solvent (). Despite the differences in the evolutionary origin (microbe, fly and human), the amino acid sequence and biophysical properties, the extent of S4 motion appears to be similar between the three channels.
Figure 4. Model of the voltage sensing paddle of hERG showing changes in accessibility to extracellular solvent during depolarization. (A) Sequence alignment of the voltage sensing paddles of the KvAP (GI: 14601099), hERG (GI: 4557729), Shaker (GI: 24642914) and bEAG (GI: 31342184) potassium channels. S3b and S4 regions are indicated by the top horizontal lines. Key voltage sensing positively charged residues are shown in bold (numbered 1–4). Residues shaded in grey are exposed to extracellular solvent in studies on Shaker [Citation17,Citation27], KvAP [Citation19,Citation26] and hERG (this study) and bEAG [Citation29]; arrow indicates the membrane-aqueous border based on the experimental data for KvAP, Shaker and hERG. (B) Structural model of the hERG paddle highlighting the residues (side chains of residues shaded grey in [A] are shown; positive charges labelled) whose position changes from being within the membrane (resting) to extracellular solvent when the channel is activated/inactivated; the dotted line denotes the putative outer membraneaqueous border for S4. The model, based on the coordinates of KvAP (PDB code: 1ORQ), is taken from the SWISS MODEL Repository [Citation32]. The paddle is orientated using the position of the S3b–S4 paddle in the Kv1.2 crystal structure (PDB code: 2A79) as a guide. It should be noted that the cartoon was not meant to indicate the type of physical motion the paddle undergoes.
![Figure 4. Model of the voltage sensing paddle of hERG showing changes in accessibility to extracellular solvent during depolarization. (A) Sequence alignment of the voltage sensing paddles of the KvAP (GI: 14601099), hERG (GI: 4557729), Shaker (GI: 24642914) and bEAG (GI: 31342184) potassium channels. S3b and S4 regions are indicated by the top horizontal lines. Key voltage sensing positively charged residues are shown in bold (numbered 1–4). Residues shaded in grey are exposed to extracellular solvent in studies on Shaker [Citation17,Citation27], KvAP [Citation19,Citation26] and hERG (this study) and bEAG [Citation29]; arrow indicates the membrane-aqueous border based on the experimental data for KvAP, Shaker and hERG. (B) Structural model of the hERG paddle highlighting the residues (side chains of residues shaded grey in [A] are shown; positive charges labelled) whose position changes from being within the membrane (resting) to extracellular solvent when the channel is activated/inactivated; the dotted line denotes the putative outer membraneaqueous border for S4. The model, based on the coordinates of KvAP (PDB code: 1ORQ), is taken from the SWISS MODEL Repository [Citation32]. The paddle is orientated using the position of the S3b–S4 paddle in the Kv1.2 crystal structure (PDB code: 2A79) as a guide. It should be noted that the cartoon was not meant to indicate the type of physical motion the paddle undergoes.](/cms/asset/3606317c-ab34-41c0-8881-a9dfc6f5d6d5/imbc_a_432282_f0004_b.jpg)
However, the situation in bEAG1 seems to be different from this common theme in that positions 320 and 321 of bEAG are inaccessible to extracellular solvent in the resting state [Citation29], whereas the corresponding positions in other channels are accessible. This may seem surprising given the fact that bEAG has a high sequence identity to hERG, but the difference may be related to the fact that bEAG has a unique Mg2+ binding site [Citation30,Citation31] that appears to modulate the extracellular crevice surrounding the S4 helix in a voltage dependent manner [Citation29].
Despite extensive studies, the mechanism of S4 movement remains highly controversial. The original observation by MacKinnon and colleagues that the structure of the paddle (S3b–S4) is conserved among all voltage sensitive Kv channels led to the suggestion that S4 does not move as an independent domain, but moves as a rigid unit with S3b, that is, as a paddle. The paddle sequence in hERG is more similar to that of KvAP than Shaker () and could be modelled onto KvAP readily (). Importantly, the loop connecting S3b–S4 is short and is of the same length (four amino acids) in hERG and KvAP. Because the loop is so short, it is difficult to envisage how a large portion of S4 (two helical turns) can move out of the bilayer without concomitant S3 motion. Thus it is likely that the S4 motion is accompanied by a simultaneous motion of S3b as suggested by some studies [Citation19,Citation20].
Evidence in favour of paddle motion is gaining support in recent years. In an elegant series of studies, Swartz group has shown that the paddles are portable: Paddles taken from voltage-gated Kv channels of diverse evolutionary origin could be functionally substituted into Kv2.1 [Citation13]. Even more striking was the finding that the paddle motif from the VSD of a non-ion channel protein, Ci-VSP, a voltage sensitive phosphatase, could be transplanted into Kv2.1 [Citation13]. In a more recent paper, the group has demonstrated that gating and pharmacological properties of paddles of voltage-gated sodium channels could be functionally transferred to Kv channels [Citation15]. Taken together, it seems that, despite large differences in the biophysical and pharmacological properties of VSDs, the molecular mechanism of voltage sensing may be conserved among all voltage sensing proteins. However, whether S4 and S3b move as a rigid S3b–S4 paddle, or move independently, remains to be determined.
Supplementary Material 1
Download JPEG Image (123.3 KB){kind=link}
Supplementary Material 2
Download JPEG Image (138.6 KB){kind=link}
Supplementary Material 3
Download JPEG Image (73.6 KB){kind=link}
Acknowledgements
This work was supported by a grant from the British Heart Foundation (PG/03/154/16411). NYD was a recipient of a visiting fellowship from the Royal Society and TUBITAK (The Scientific and Technical Research Council of Turkey).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. 1995. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95.
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. 1995. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307.
- Sanguinetti MC, Tristani-Firouzi M. 2006. hERG potassium channels and cardiac arrhythmia. Nature 440:463–469.
- Perrin MJ, Subbiah RN, Vandenberg JI, Hill AP. 2008. Human ether-a-go-go related gene (hERG) K+ channels: Function and dysfunction. Prog Biophys Mol Biol 98:137–148.
- Smith PL, Baukrowitz T, Yellen G. 1996. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379:833–836.
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. 1996. Fast inactivation causes rectification of the IKr channel. J Gen Physiol 107:611–619.
- Schönherr R, Heinemann SH. 1996. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol 493:635–642.
- Bezanilla F. 2008. How membrane proteins sense voltage. Nat Rev Mol Cell Biol 9:323–332.
- Swartz KJ. 2008. Sensing voltage across lipid membranes. Nature 456:891–897.
- Tombola F, Pathak MM, Isacoff EY. 2006. How does voltage open an ion channel? Annu Rev Cell Dev Biol 22:23–52.
- Long SB, Campbell EB, Mackinnon R. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309:897–903.
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. 2003. X-ray structure of a voltage-dependent K+ channel. Nature 423:33–41.
- Alabi AA, Bahamonde MI, Jung HJ, Kim JI, Swartz KJ. 2007. Portability of paddle motif function and pharmacology in voltage sensors. Nature 450:370–375.
- Tao X, MacKinnon R. 2008. Functional analysis of Kv1.2 and paddle chimera Kv channels in planar lipid bilayers. J Mol Biol 382:24–33.
- Bosmans F, Martin-Eauclaire MF, Swartz KJ. 2008. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456:202–208.
- Larsson HP, Baker OS, Dhillon DS, Isacoff EY. 1996. Transmembrane movement of the shaker K+ channel S4. Neuron 16:387–397.
- Yusaf SP, Wray D, Sivaprasadarao A. 1996. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflügers Arch 433:91–97.
- Yang N, George ALJr., Horn R. 1996. Molecular basis of charge movement in voltage-gated sodium channels. Neuron 16:113–122.
- Ruta V, Chen J, MacKinnon R. 2005. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell 123:463–475.
- Elliott DJ, Neale EJ, Aziz Q, Dunham JP, Munsey TS, Hunter M, Sivaprasadarao A. 2004. Molecular mechanism of voltage sensor movements in a potassium channel. EMBO J 23:4717–4726.
- Zhang M, Liu J, Tseng GN. 2004. Gating charges in the activation and inactivation processes of the HERG channel. J Gen Physiol 124:703–718.
- Smith PL, Yellen G. 2002. Fast and slow voltage sensor movements in HERG potassium channels. J Gen Physiol 119:275–293.
- Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M. 2003. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci USA 100:10534–10539.
- Zhang M, Liu J, Jiang M, Wu DM, Sonawane K, Guy HR, Tseng GN. 2005. Interactions between charged residues in the transmembrane segments of the voltage-sensing domain in the hERG channel. J Membrane Biol 207:169–181.
- Baker OS, Larsson HP, Mannuzzu LM, Isacoff EY. 1998. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+; channel gating. Neuron 20:1283–1294.
- Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. 2003. The principle of gating charge movement in a voltage-dependent K+ channel. Nature 423:42–48.
- Wang MH, Yusaf SP, Elliott DJ, Wray D, Sivaprasadarao A. 1999. Effect of cysteine substitutions on the topology of the S4 segment of the Shaker potassium channel: Implications for molecular models of gating. J Physiol 521:315–326.
- Neale EJ, Elliott DJ, Hunter M, Sivaprasadarao A. 2003. Evidence for intersubunit interactions between S4 and S5 transmembrane segments of the Shaker potassium channel. J Biol Chem 278:29079–29085.
- Schönherr R, Mannuzzu LM, Isacoff EY, Heinemann SH. 2002. Conformational switch between slow and fast gating modes: Allosteric regulation of voltage sensor mobility in the EAG K+ channel. Neuron 35:935–949.
- Silverman WR, Bannister JP, Papazian DM. 2004. Binding site in eag voltage sensor accommodates a variety of ions and is accessible in closed channel. Biophys J 87:3110–3121.
- Liu J, Zhang M, Jiang M, Tseng GN. 2003. Negative charges in the transmembrane domains of the HERG K channel are involved in the activation and deactivation-gating processes. J Gen Physiol 121:599–614.
- Kopp J, Schwede T. 2006. The SWISS-MODEL Repository: New features and functionalities. Nucleic Acids Res 34:D315–318.