Abstract
The serotype-specific glucosyltransferase, GtrV, is responsible for glucosylation of the O-antigen repeating unit of Shigella flexneri serotype 5a strains. GtrV is an integral inner membrane protein with two essential periplasmic loops: the large Loop 2 and the C-terminal Loop 10. In this study, the full length of the Loop 2 was shown to be necessary for GtrV function. Site-directed mutagenesis within this loop revealed that conserved aromatic and charged amino acids have a critical role in the formation of the active site. Sequential deletions of the C-terminal end indicated that this region may be essential for assembly of the protein in the cytoplasmic membrane. The highly conserved FWAED motif is thought to form the substrate-binding site and was found to be critical in GtrV and GtrX, a serotype-specific glucosyltransferase with homology to GtrV. The data presented constitutes a targeted analysis of the formation of the GtrV active site and highlights the essential role of the large periplasmic Loop 2 in its function.
Introduction
Shigella spp. are the causative agents of shigellosis or bacillary dysentery. S. flexneri is the most prevalent species in less developed countries and accounts for more disease worldwide than any other dysentery-causing strain of E. coli (Kotloff et al. Citation1999). In Shigella spp., the O-antigen is the most variable part of the lipopolysaccharide (LPS) and is highly immunogenic (Liu et al. Citation2008). The O-antigen of S. flexneri, with the exception of serotype 6 strains, consists of several repeating units of a basic tri-rhamnose-N-acetylglucosamine tetrasaccharide (Van den Bosch et al. Citation1997, Allison & Verma Citation2000). In S. flexneri serotype 5a strains, the basic O-antigen repeating unit is glucosylated by a phage-encoded protein called GtrV. This glucosyltransferase is an inner membrane integral protein thought to be responsible for the periplasmic transfer of a lipid phosphate-activated glucosyl residue (UndP-glucose) to rhamnose II of the O-antigen repeating unit. Similarly, GtrX is a phage-encoded protein of S. flexneri serotype X strains involved in the specific attachment of a glucosyl residue to rhamnose I of the basic tetrasaccharide.
Topology studies of GtrV revealed that it has a cytoplasmic N-terminus, nine transmembrane helices, a hypothetical re-entrant loop between the fourth and fifth transmembrane regions and a C-terminal periplasmic end (Korres & Verma Citation2004). Previous functional studies in GtrV reported a large periplasmic Loop 2 between the first and second transmembrane regions, and a long C-terminal Loop 10, as essential for GtrV enzymatic activity (Korres & Verma Citation2006). Furthermore, three critical acidic residues, E42 and D43 in Loop 2 and D380 in Loop 10, were identified (Korres & Verma Citation2006). In the same study, Korres and colleagues hypothesized that the periplasmic Loop 2 is involved in the interaction with the undecaprenol (UndP)-glucose precursor, and the C-terminal Loop 10 is responsible for the specific attachment of the glucosyl residue to the O-antigen repeating unit. This was reasoned since chimeras constructed between GtrV and GtrX consisting of GtrV backbone with the Loop No 2 of GtrV swapped with Loop No 2 of GtrX maintained serotype conversion to 5a. This indicated that Loop No 2 of GtrX was functioning in a similar manner to Loop No 2 of GtrV primarily due to the conserved interaction with UndP-glucose precursor. Based on this hypothesis, the general aim of this work was to identify critical residues or motifs in the large Loop 2 and in the C-terminal Loop 10 for further clues on the active site of GtrV. This study included deletion of amino acids segments and replacement of specific amino acids in GtrV and GtrX and revealed that the catalytic function of GtrV appears to be more centred in the large periplasmic Loop 2, suggesting a similar scenario for other inner membrane multispanning transmembrane glycosyltransferases.
Materials and methods
Bacterial strains and growth conditions
E. coli XL1-Blue MRF' [Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac] was used for construction and maintenance of plasmids. E. coli JM110 [rpsL (Strr) thr leu thi-1 lacY galK galT ara tonA tsx dam dcm supE44 Δ(lacproAB)] was used to prepare plasmid DNA free of Dam and Dcm methylation. S. flexneri strain SFL1616 (auxotrophic serotype Y strain carrying a pACYC177-based plasmid encoding GtrA and GtrB) was used for expression of the recombinant proteins (Lehane et al. Citation2005). Bacteria were cultured under aerobic conditions in Luria-Bertani (LB) media at 37°C supplemented with the appropriate antibiotics. Alkaline phosphatase and β-galactosidase activities were detected by the blue or red colour formation, respectively, of colonies on dual indicator (DI) agar plates containing antibiotics and 80 μg/ml of 5-bromo-4-chloro-3-indolyl phosphatise disodium salt (X-phos), 100 μg/mL of 6-chloro-3-indolyl-β-D-galactoside (Red-Gal), 1mM IPTG and 80 mM K2HPO4 (pH 7.0). Antibiotics were used at the following concentrations: ampicilin (Ap), 100 μg/ml; chloramphenicol (Cm), 25 μg/ml; tetracycline (Tc), 12.5 μg/ml; kanamycin (Km), 50 μg/ml.
Plasmids and general DNA manipulation procedures
Standard methods were performed, if not otherwise indicated, according to Sambrook and Russell (Citation2001). Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen). Restriction enzymes (Fermentas) and other nucleic-acid-modifying enzymes such as T4 DNA ligase (Promega) and Klenow fragment (Fermentas) were used as recommended by the manufacturers' instructions. In vitro amplification of DNA by PCR was performed using primers purchased from Sigma-Aldrich and the high fidelity DNA polymerase PfuUltra II (Stratagene). All the constructs were confirmed to be error-free by sequencing at the Biomolecular Resources Facility, John Curtin School of Medical Research (Australian National University, Canberra) using the Big Dye Version 3.1 protocol. For full details on plasmids and oligonucleotide primers used in this study see Supplementary Tables I and II, respectively (online version only).
Construction of gtrV-phoA-lacZα translational fusions
For GtrVWT-PhoA-LacZα constructs, gtrV was amplified from pNV323 (Supplementary Table I, Panel A) and cloned into a SacI/StuI ∼4.87 kb digested fragment of pNV1090 (Supplementary Table I, Panel A), a pBC SK-based plasmid that harbours the pho-lac dual reporter created by Alexeyev & Winkler (Alexeyev & Winkler Citation1999). For gtrV amplification, the 5′ primer (GtrVFSacI; Supplementary Table II, Panel A) was designed to include 12 bp upstream of the ATG start codon of gtrV, while the 3′ primer (GtrVRStuI; Supplementary Table II, Panel A) was designed to match the C-terminal region of GtrV in a way that leaves out the last P417 residue and the TAA stop codon. The construct produced, plasmid pNV1708, was checked for precise in-frame fusion by sequence analysis and used as template for site-directed mutagenesis studies in GtrV. Plasmid pNV1709 is a HindIII-, EcoRV- and NruI restriction sites-free version of pNV1708 created by blunt-end ligation of a HindIII/NruI digested pNV1708.
Construction of in-frame GtrV Loop 2 deletions
In-frame deletions of the periplasmic Loop 2 of GtrV were generated by PCR. Plasmid pNV1709, containing the coding sequence for a GtrVWT-PhoA-LacZα fusion protein was used as template. Primers were designed to incorporate NruI restriction sites (Supplementary Table II, Panel B) and six PCR products were generated in order to obtain the sequential Loop 2 deletions R30 to P37, Q38 to H45, V46 to N54, I56 to P63, P63 to K73 and S77 to Y86 (designated L2Δ1 to L2Δ6, respectively). After NruI restriction digest and ligation, plasmids (listed on Supplementary Table I, Panel B) were checked for in-frame junction of the 5′ and 3′ ends by sequencing.
Construction of GtrV C-terminal truncations
PCR amplification was used to generate constructs encoding C-terminal truncations of GtrV with deletion of 5, 9, 12 and 19 residues from the C-terminal end (designated L10Δ1 to L10Δ4, repectively). Plasmid pNV1105 (encoding GtrVL10Δ1-PhoA-LacZα) was previously obtained by in-frame fusion of the dual reporter pho-lac to residue I413 of GtrV (Korres & Verma Citation2004). pNV1646 (GtrVL10Δ2) and pNV1627 (GtrVL10Δ4) were created by introduction of a new termination TAA codon at the desired positions T409 and Y399, respectively. Plasmid pNV1805 (GtrVL10Δ3-PhoA-LacZα) was created by in-frame fusion of the reporter pho-lac to P405. For full details, see Supplementary Tables I and II, Panel C. In-frame fusion to pho-lac and stop codon insertion by site-directed mutagenesis was confirmed by sequence analysis.
Site-directed mutagenesis on Loop 2 and Loop 10
Specific individual and pair amino acid mutations of both GtrV and GrtX were generated by site-directed mutagenesis using the method based on the QuickChange Site-Directed Mutagenesis Kit (Stratagene) and DNA polymerase PfuUltra II. The cognate oligonucleotides used are listed in Supplementary Table II, Panel D and E. Presence of the mutations in the plasmids was verified by sequence analysis. All the created constructs together with their relevant features are listed in Supplementary Table I, Panel D and E.
In vivo determination of the activity of GtrV and GtrX derivatives
The S. flexneri strain SFL1616 (serotype Y) created by Lehane et al. (Citation2005) was used as the host for functional determination of the various GtrV and GtrX derivatives. Slide agglutination test and colony and LPS Western immunoblots were used to observe whether the host serotype Y strain (group antigens 3 and 4) was converted to 5a (type antigen V) or X (group antigens 7 and 8) serotypes upon expression of plasmid-encoded GtrV or GtrX derivatives, respectively. Monovalent antisera group (3)4, type V and group 7(8) from Denka Seiken was used. For immunoblots, horseradish peroxidise-conjugated goat anti-rabbit IgG (Sigma) was used as secondary antibody and binding was detected using the SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo). On LPS Western immunoblots, the volume of culture harvested was equal to 0.55/OD600 and the SDS-proteinase K whole-cell lysate samples were made following the procedure of Hitchcock & Brown (Hitchcock & Brown Citation1983).
Bioinformatic analysis
Accession numbers of the nucleotide and amino acid sequences used are listed in Supplementary Table III (online version only). Pairwise comparison of GtrV and GtrX with other proteins in the GenBank were performed using the NCBI BLAST program (Johnson et al. Citation2008). Multiple alignments between GtrV, GtrX or other sequences were performed using ClustalW (Larkin et al. Citation2007). Hydrophobicity plots were obtained using the DAS (Cserzo et al. Citation1997) and TMpred (Hofmann Citation1993) programs. Similarity and identity scores were obtained using MatGAT (Campanella et al. Citation2003).
Results
GtrVWT-PhoA-LacZα translational fusions are functional
GtrV is a 417 amino acid-long transmembrane protein responsible for the glucosylation of rhamnose II of the S. flexneri O-antigen repeating unit (Allison & Verma Citation2000). Fusions of this protein to the pho-lac dual reporter (Alexeyev & Winkler Citation1999), consisting of both E. coli alkaline phophatase gene (phoA) and α-fragment of the β-galactosidase gene (lacZα), were successfully used by Korres and Verma (Citation2004) to present a topology model for GtrV. In this study, fusion of the reporter to the periplasmic C-terminal end of GtrV (GtrVWT-PhoA-LacZα) was used as the starting material, thus allowing the GtrV derivatives to be assayed for synthesis and assembly in the inner membrane of the bacterial cell envelope. In order to test if the GtrVWT-PhoA-LacZα translational fusion was functional, plasmids pNV1708 and pNV1709 (Supplementary Table I) were transformed into serotype Y electrocompetent SFL1616 cells. Slide agglutination tests and LPS immunoblots showed that both plasmids were able to confer the serotype 5a phenotype to the serotype Y strain (SFL1616) (data not shown), suggesting that the GtrVWT-PhoA-LacZα translational fusion was functional. In addition to the functional assay, blue coloration was observed when the constructs were transformed into XL1-Blue MRF' electrocompetent cells and patched onto DI plates (data not shown). Taken together, these results suggest that fusion of PhoA-LacZα to GtrV does not interfere with its catalytic activity and further confirms the periplasmic position of the C-terminal end of GtrV.
Periplasmic Loop 2 is essential for in vivo GtrV activity
GtrV spans the inner membrane nine times and has the N-terminus located in the cytoplasm whereas the C-terminus is in the periplasm (Korres & Verma Citation2004). The periplasmic Loop 2 is thought to be of functional importance. In order to investigate its role, a series of constructs were created encoding GtrV mutants with sequential in-frame Loop 2 deletions (L2Δ1 to L2Δ6) (). These deletions were designed to divide Loop 2 into six different segments with an approximate length of 9 amino acids each.
Figure 1. Functional characterization of GtrV Loop 2 deletions. (A) Schematic representation of the segments deleted within periplasmatic Loop 2 of GtrV (yellow). The first and last residues of each deleted segment are represented. Note that GtrVWT-PhoA-LacZα was used as template and that the fused dual reporter proteins are not represented. (B) The positive control (+ control) corresponds to the signal given by pNV1709 in SFL1616 cells. The negative control (- control) corresponds to the signal given by SFL1616. LPS was detected by Western immunoblotting using type V antisera. Slide agglutination results using type V antisera are indicated by (+) and (-) according to occurrence or not of agglutination, respectively. (C) Coloration of XL1-Blue MRF' recombinant colonies when patched onto DI plates. Blue is indicative of protein assembly. Results from (B) and (C) are representative of the phenotypes observed in at least three independent experiments. (This Figure is reproduced in colour in Molecular Membrane Biology online.)
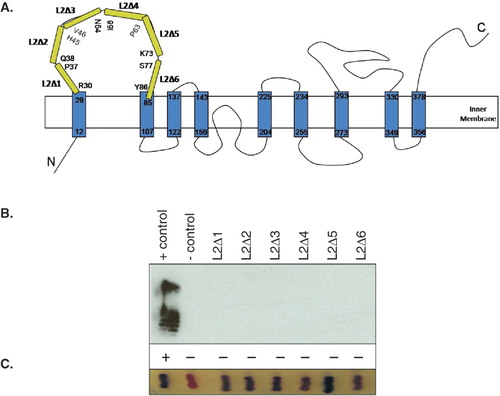
In vivo GtrV activity was assessed by LPS immunoblot using type V antisera and showed positive reaction for the expression of GtrVWT-PhoA-LacZα. The same was not true, however, for all the in-frame GtrV Loop 2 deletions (). The six mutant proteins although being synthesized and assembled in the membrane, as indicated by the blue colour produced by the exposure of the PhoA to the periplasm (), were unable to mediate serotype conversion. This means that the activity of the GtrV derivatives was compromised indicating that deletion of segments of the loop may either have affected local protein folding or that each deleted segment contained residues essential for the GtrV catalytic activity.
Similarities shared by GtrV and other proteins
Among all the S. flexneri serotype-specific glucosyltransferases, the primary sequences of GtrV (from serotype 5a strains) and GtrX (serotype X strains) share some degree of homology (34.2% identity and 58.8% similarity) and highly conserved domains, especially in the N-terminal half of the protein, are found. For further analysis of the conserved regions of GtrV and GtrX, BLAST searches were performed. A number of sequences sharing homology with both proteins were identified (Supplementary Table IV –online version only). Three of them, belonging to the complete sequenced genomes of Anabaena variabilis (YP_323852), Granulibacter bethesdensis (YP_744562) and Burkholderia phytofirmans (YP_001894533) were considered of greatest interest for this study (Supplementary Figure I – online version only). According to hydrophobicity data generated from the amino acid sequences using DAS (Cserzo et al. Citation1997) and TMpred (Hofmann Citation1993) programs, the proteins appear to be transmembrane proteins and to contain 8–10 multiple hydrophobic regions (data not shown). Alignment of the N-terminal region of GtrV, GtrX and these other proteins demonstrates the presence of highly conserved residues and motifs in the Loop 2 region ().
Figure 2. Identification of conserved residues in Loop 2 of GtrV and functional characterization. (A) GtrV, GtrX and the first three proteins of Supplementary Table IV were aligned using ClustalW (Larkin et al. Citation2007). The identical amino acids are highlighted in grey boxes; dashed boxes indicate conserved substitutions. The first two transmembrane segments of GtrV are indicated in the bottom of each line by filled bars. The previously identified E42 and D43 critical residues (Korres & Verma Citation2006) are underlined. Asterisks indicate the residues that were subjected to site-directed mutagenesis in GtrV. (B) Functional characterization of GtrV-PhoA-LacZα derivatives carrying mutation of Loop 2 residues. Plasmids were transformed into electrocompetent SFL1616 cells. The positive control (+ control) corresponds to the signal given by pNV1708 in SFL1616 cells. The negative control (- control) corresponds to the signal given by SFL1616. LPS was detected by Western immunoblotting using type V antisera. Slide agglutination results using type V antisera are indicated by (+) and (-) according to occurrence or not of agglutination, respectively. (C) Coloration of XL1-Blue MRF' recombinant colonies on DI plates. Blue is indicative of protein assembly. Results from (B) and (C) are representative of the phenotypes observed in at least three independent experiments. (This Figure is reproduced in colour in Molecular Membrane Biology online.)
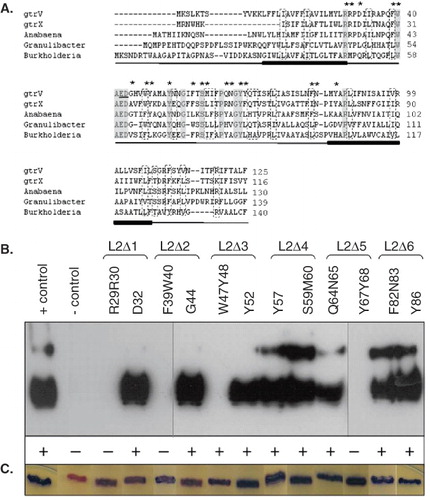
Identification of critical residues in Loop 2 of GtrV
The assumption that each of the deleted segments potentially encompasses critical residues was tested by site-directed mutagenesis. Target residues were chosen on the basis of their chemical properties and the conservation between the sequences of GtrV, GtrX and other putative glucosyltransferases. Multiple alignments of GtrV and GtrX with the highest scored proteins of a BLAST search (Supplementary Table IV and Supplementary Figure I) allowed the identification of several conserved motifs, most of them containing aromatic and/or charged residues (), which are known to be more likely involved in the interaction with monosaccharides (Garinot-Schneider et al. Citation2000, Marks et al. Citation2001, Crouvoisier et al. Citation2007). Overall, 12 mutations within Loop 2 were carried out. These included modification to alanine of individual and pairs of amino acids. Residues R29R30 and D32 were mutated within L2Δ1 segment, F39W40 and G44 within L2Δ2, W47Y48 and Y52 within L2Δ3, F57 and S59M60 within L2Δ4, Q64N65 and Y67Y68 within L2Δ5 and finally, F82N83 and Y86 within L2Δ6.
All mutant proteins were synthesized and assembled in the membrane, as assessed by patching transformed XL1-Blue MRF' cells onto DI plates (). Loss or presence of enzyme activity of the GtrV mutated proteins expressed in SFL1616 and immunodetection of serotype conversion is presented in . Expression of the GtrV derivatives carrying either mutations R29R30, F39W40 or Y67Y68 were unable to change the SFL1616 phenotype. These observations imply that the mutated residues play a critical role in protein function. Point mutations of the individual amino acid residues in the above pairs revealed that R29 and Y67 were solely responsible for the observed loss of function while in the case of the F39W40 mutant both residues appeared to be critical for the function of GtrV. In the case of the aromatic pair W47Y48, a residual enzyme activity was detected by slide agglutination test (). In contrast to the mutation of conserved positively charged and/or aromatic amino acid pairs, mutation of an unconserved aromatic residue (F57) and of positively charged amino acids surrounded by neutral amino acids (Y52 and Y86) led to the conversion of the SFL1616 phenotype. Similarly, mutation of the conserved pair S59M60 did not have an effect on GtrV function, as well as mutations F82N83 (carrying an aromatic and conserved amino acid) and Q64N65 (carrying a pair of polar residues). When the conserved negatively charged residue D32 was mutated, the expressed protein remained functional. This also holds true for the mutation of the unconserved, uncharged G44 residue. In summary, GtrV critical residues were identified in segments L2Δ1, L2Δ2 and L2Δ5. In all these cases the targeted residues belonged to conserved pairs of aromatic or positively charged amino acids. In L2Δ3, a pair of aromatic amino acids compromised the activity of GtrV. Residues affecting function of GtrV failed to be identified in segments L2Δ4 and L2Δ6.
Identification of critical residues in GtrX
GtrX is a serotype-specific glucosyltransferase present in S. flexneri serotype X strains. It catalyses the same reaction as GtrV but the acceptor molecule for the glucosyl group is rhamnose I of the O-antigen repeating unit. Due to the wide diversity of acceptor molecules, glucosyltransferases have generally very low sequence homology but similarity between GtrV and GtrX is evident at both primary and secondary structures level.
To study the function of GtrX and investigate its similarity to GtrV, equivalent amino acids known to be critical residues in GtrV were targeted and changed to alanine in GtrX. Specifically, mutation of the conserved, charged and/or aromatic amino acid pairs R20R21 (corresponding to the critical R29R30 pair in GtrV), F30W31 (corresponding to F39W40), E33D34 (corresponding to E42D43) and W38Y39 (corresponding to W47Y48) was undertaken. For comparison purposes, residue F57 shown not to be critical in GtrV and corresponding to F48 in GtrX was also mutated. Site-directed mutagenesis was performed on the pBC SK-based plasmid pNV1057 carrying the full length gtrX (Supplementary Table I, Panel A) and resulted in the creation of five GtrX mutants. S. flexneri serotype Y strain SFL1616 was used as the host for functional characterization. The results (data not shown) showed that mutation of the equivalent GtrV F48 non-critical residue did not compromise GtrX activity while mutation of the corresponding GtrV critical residues R20R21, F30W31 and E33D34 abolished function of GtrX. The pair W38Y39, which compromised GtrV catalytic activity, rendered GtrX non-functional. The GtrX gene was not fused with the pho-lac dual reporter, thus the expression and assembly of the protein in the membrane could not be monitored. However, the consistency of these results with the results seen for GtrV allows their correlation to the loss of GtrX enzymatic activity. Moreover, they reflect the strong relation existing between the structure and function of these proteins and constitute a direct evidence of a common and conserved role for the large periplasmic Loop 2 of GtrV.
Importance of the C-terminal region of GtrV
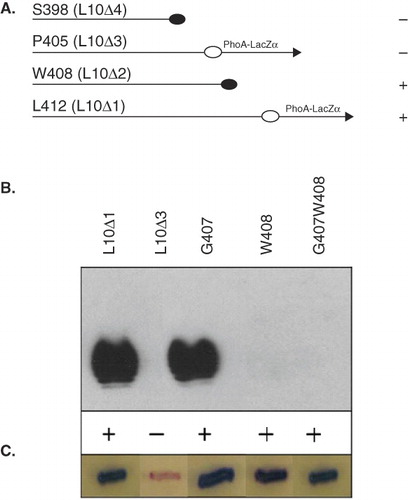
The C-terminal Loop 10 is the second biggest periplasmic segment of GtrV and has been reported to be of functional importance (Korres & Verma Citation2006). While the large periplasmic Loop 2 is thought to interact with the donor substrate, the C-terminal region is probably involved in the specific attachment of the glucosyl residue to the O-antigen repeating unit. The function of the C-terminal region was investigated through a similar approach to the periplasmic Loop 2. Deletion mutants of 5, 9, 12 and 19 residues from the C-terminus of GtrV were created (L10Δ1 to L10Δ4) and functionality was assessed by slide agglutination test and LPS immnublotting. As shown in , deletion of 5 and 9 residues did not abolish function of GtrV but deletion of 12 and 19 residues caused the protein to be nonfunctional. These results suggest that almost the intact length of the C-terminal region is essential for GtrV function, but the red coloration produced by the GtrV mutant lacking the last 12 residues (L10Δ3) () reveals that the protein was not able to correctly reach its destination. To further elucidate whether the 12 amino acid-long deleted segment (L10Δ3) was relevant to GtrV activity, a site-directed mutagenesis approach was used. The GtrV derivative lacking the last nine residues (L10Δ2) was capable of converting serotype Y to 5a so it was speculated that (i) it is unlikely that critical residues lie in this portion of the C-terminal region and (ii) if any of the 12 last residues are important for GtrV function, they are present between L10Δ3 and L10Δ2. Hence, only one or a combination of residues A406, G407 and W408 can potentially be critical for GtrV activity and alanine substitution of G407, W408 and G407W408 was performed. Functional assays indicated that amino acid W408 alone, or together with the adjacent G407, compromises GtrV activity but is incapable of rendering the protein non-functional, as indicated by the positive reaction of the agglutination test ().These results indicate that the residue W408 is critical for GtrV function and the C-terminus Loop 10 may be important for the stability and/or membrane assembly of GtrV, but this has to be explored further.
Figure 3. Functional characterization of the C-terminal region of GtrV. Signals (+) and (-) indicate occurrence or not of agglutination with type V antisera. Plasmids were transformed into electrocompetent SFL1616 cells. (A) Diagram showing the different length of the C-terminal truncations and the ability of the GtrV derivatives to mediate serotype conversion. Constructs are represented by the C-terminal region. Truncations are designated by the last GtrV amino acid before the PhoA-LacZα fusion (arrow) or stop codon incorporation (filled circles). (B) LPS detected by Western immunoblotting and agglutination results using type V antisera. (C) Coloration of XL1-Blue MRF' recombinant colonies on DI plates; blue is indicative of protein assembly. Results from (B) and (C) are representative of the phenotypes observed in at least three independent experiments. (This Figure is reproduced in colour in Molecular Membrane Biology online.)
Discussion
Structural and functional analysis of proteins is carried out to understand how different parts of a polypeptide chain contribute to the form and activity of the whole molecule (Alexeyev & Winkler Citation2002). Mutagenesis studies are one of the approaches that can be used in such an analysis and may include amino acid replacement and deletion or insertion of different length amino acid segments (Alexeyev & Winkler Citation2002). Although the evidence is indirect, glucosylation of the O-antigen by GtrV and GtrX is thought to take place at the periplasmic side of the inner membrane (Guan et al. Citation1999; Korres et al. Citation2005). The topology of GtrV was shown to include two large and essential periplasmic loops: the 55 amino acid-long Loop 2 in the N-terminal region and the 39 amino acid-long C-terminal Loop 10 (Korres & Verma Citation2006). The existence of large periplasmic loops is seen in several proteins whose activity is known to occur in the periplasmic side of the membrane. For instance, the S. flexneri O-antigen polymerase Wzy has been biochemically proven to act at the periplasmic side and was shown by Daniels et al. (Citation1998) to have 12 transmembrane helices and two large periplasmic segments (53 and 21 amino acids). In previous work, it was speculated that in both GtrV and GtrX, the conserved periplasmic Loop 2 is involved in the interaction with the donor sugar UndP-glucose and that the relatively large C-terminal Loop 10 is required for the attachment of the glucosyl residue to the O-antigen repeating unit (Korres & Verma Citation2006). Herein, functional analysis of GtrV was carried out by deletion of periplasmic segments of the protein and by introduction of several point mutations. Functional characterization of GtrV and GtrX was assessed by the in vivo ability of plasmid-encoded glucosyltransferases to mediate serotype conversion of the attenuated Y strain SFL1616. As the fusion of the pho-lac dual reporter to GtrV did not seem to affect its catalytic property, plasmids encoding GtrVWT-PhoA-LacZα translational fusions were shown to be suitable in the study of GtrV function. Their derivatives allowed determination of whether the loss of function was a consequence of (i) deletion/mutation of critical segments/residues or alternatively, (ii) the production of unstable or misfolded proteins.
Sequential in-frame deletions of Loop 2 in GtrV showed that the full length of the loop is essential for enzyme activity but not for assembly of the protein in the membrane (). This evidence implicates the periplasmic Loop 2 in the formation of the active site and is in good agreement with previous literature where even conservative substitutions of residues within this region (E42 and D43) resulted in loss of GtrV activity (Korres & Verma Citation2006). Since all the six GtrV deletion derivatives were non-functional, it was speculated that each deleted segment may contain critical residues for GtrV activity or, additionally, may have residues that participate in the proper folding of the loop or that are indirectly necessary for the interaction between GtrV and its donor sugar.
Individual and pairs of amino acids within Loop 2 were investigated. The S. flexneri bacteriophage-encoded glucosyltransferases identified to date, with the exception of GtrV and GtrX, share little sequence homology and provide limited information on the identification of conserved residues. However, BLAST searches identified a number of proteins with homology to GtrV and GtrX (Supplementary Table IV) that allowed the identification of several conserved motifs within the Loop 2 region, most of them enclosing aromatic and/or charged residues ().
Conserved motifs in Loop 2 were found to be critical not only in GtrV but also in GtrX. Site-directed mutagenesis carried out in GtrV attributed the non-functionality of three of the six Loop 2 deletion mutants (L2Δ1, L2Δ2 and L2Δ5), to the presence of critical residues. Segment L2Δ3 was shown to contain an aromatic motif that compromises GtrV activity and abolishes GtrX function. No critical residues were found in deleted segments L2Δ4 and L2Δ6. These segments, together with L2Δ1 and L2Δ3, may be necessary to bring L2Δ2 and L2Δ5 in close proximity, thus allowing the formation of the substrate-binding site or, broadly, of the protein active site.
In addition to the previously identified negatively charged amino acid pair E42D43 (Korres & Verma Citation2006), amino acids R29, F39, W40 and Y67 were crucial for GtrV activity. The equivalent pairs E33D34, R20R21 and F30W31, in addition with pair W38Y39, were crucial for GtrX activity. All the identified amino acid pairs are conserved aromatic or charged residues, emphasizing the relevance of these amino acids in the formation of protein active sites. Not all the mutations within the periplasmic Loop 2 were able to abolish function of GtrV or GtrX, which means that although the full length of the loop is necessary for function, only specific residues define essential components of the GtrV and GtrX active site.
The combination of topological evidence with the sequence analysis and genetic data generated in this study, allows the prediction of a putative folding pattern for Loop 2. The pairs F39W40 and E42D43 (numbered according to GtrV sequence) are part of a highly conserved motif (FWAED motif) and are thought to be directly responsible for the interaction with the donor sugar. The R29 residue, in contrast, is probably necessary to make the FWAED motif an exposed region of the loop due to its localization in the interface of the first transmembrane region with the periplasm. The W38Y39 motif identified as critical in GtrX, is positioned just after the highly conserved FWAED motif and before an 18-amino acid-long segment where no critical residues were identified. In this segment, two highly conserved glycine residues exist and in a rat glucosylceramide synthase, glycine residues have been shown to be critical for the folding of the protein and thought to have a similar role in other glycosyltransferases (Marks et al. Citation2001). Therefore, it seems reasonable to think that W38Y39 motif is critical for the proper folding of the loop, maybe allowing the 18-amino acid-long segment to interact and consequently, bringing the FWAED motif close to the critical Y67 residue.
The function of the C-terminal region was also investigated (). Although previously it was suggested that the C-terminal region of GtrV has a specific role in catalysis, none of the results of this study can undoubtedly establish that. First, deletion of the C-terminal region seems to have a stronger impact on the stability or assembly of the protein than in its function. Second, mutation of W408 to alanine seems to be insufficient to abolish GtrV activity. Finally, the previous mutation of D380 only rendered GtrV non-functional after alanine substitution (Korres & Verma Citation2006). Substitution of the aspartic acid residue to asparagine was unable to abolish the protein activity and if this residue was a critical component of the protein active site, then even the slightest variation in the chemical properties of the residue would have caused a complete loss of GtrV activity.
In summary, by mutagenesis studies and in vivo analysis an improved understanding of the structure and function of the serotype-specific glucosyltransferase GtrV has been achieved. Deletion of Loop 2 segments showed that the full length of the loop is necessary for the GtrV function. A critical highly conserved FWAED motif was identified in Loop 2 and is thought to be directly responsible for the formation of the active site, most probably the substrate-binding site. Additional critical motifs found within Loop 2 of GtrV were conserved aromatic or charged amino acids. A conserved mode of action between GtrV and GtrX is suggested by the finding that critical motifs within Loop 2 of GtrV were also critical in GtrX. Alignements between GtrV, GtrX and three putative glucosyltransferases identified by BLAST searches showed that conservation of GtrV decreases towards the C-terminal region. In this region, deletion of 12 residues caused an improper assembly of the protein in the membrane and mutation of a tryptophan residue in the full length GtrV compromised its activity. Although the present analysis gives an insight into a possible folding pattern for Loop 2, the precise catalytic mechanism of the protein is still unknown and requires further crystallographic and enzymological study.
Supplementary Material
Download PDF (257.8 KB)Acknowledgements
JAM thanks F. Tavares for constant support during the Masters program.
Declaration of interest: This work was supported by a grant from the National Health and Medical Research Council of Australia (Grant number 366743). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alexeyev MF, Winkler HH. 1999. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J Mol Biol 285(4):1503–1513.
- Alexeyev MF, Winkler HH. 2002. Transposable dual reporters for studying the structure-function relationships in membrane proteins: permissive sites in R. prowazekii ATP/ADP translocase. Biochemistry 41(1):406–414.
- Allison GE, Verma NK. 2000. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol 8(1):17–23.
- Campanella JJ, Bitincka L, Smalley J. 2003. MatGAT: an application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics 4:29.
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 22:480–484.
- Crouvoisier M, Auger G, Blanot D, Mengin-Lecreulx D. 2007. Role of the amino acid invariants in the active site of MurG as evaluated by site-directed mutagenesis. Biochimie 89(12):1498–1508.
- Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A. 1997. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein engineering 10(6):673–676.
- Daniels C, Vindurampulle C, Morona R. 1998. Overexpression and topology of the Shigella flexneri O-antigen polymerase (Rfc/Wzy). Mol Microbiol 28(6):1211–1222.
- Garinot-Schneider C, Lellouch AC, Geremia RA. 2000. Identification of essential amino acid residues in the Sinorhizobium meliloti glucosyltransferase ExoM. J Biol Chem 275(40):31407–31413.
- Guan S, Bastin DA, Verma NK. 1999. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology 145(Pt 5):1263–1273.
- Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154(1):269–277.
- Hofmann K, and W. Stoffel. 1993. TMbase – A database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler 374:166.
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res 36(Web Server issue):W5–9.
- Korres H, Mavris M, Morona R, Manning PA, Verma NK. 2005. Topological analysis of GtrA and GtrB proteins encoded by the serotype-converting cassette of Shigella flexneri. Biochem Biophys Res Commun 328(4):1252–1260.
- Korres H, Verma NK. 2004. Topological analysis of glucosyltransferase GtrV of Shigella flexneri by a dual reporter system and identification of a unique reentrant loop. J Biol Chem 279(21):22469–22476.
- Korres H, Verma NK. 2006. Identification of essential loops and residues of glucosyltransferase V (GtrV) of Shigella flexneri. Mol Membr Biol 23(5):407–419.
- Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77(8):651–666.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948.
- Lehane AM, Korres H, Verma NK. 2005. Bacteriophage-encoded glucosyltransferase GtrII of Shigella flexneri: membrane topology and identification of critical residues. Biochem J 389(Pt 1):137–143.
- Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, Reeves PR, Wang L. 2008. Structure and genetics of Shigella O antigens. FEMS Microbiol Rev 32(4):627–653.
- Marks DL, Dominguez M, Wu K, Pagano RE. 2001. Identification of active site residues in glucosylceramide synthase. A nucleotide-binding catalytic motif conserved with processive beta-glycosyltransferases. J Biol Chem 276(28):26492–26498.
- Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Van den Bosch L, Manning PA, Morona R. 1997. Regulation of O-antigen chain length is required for Shigella flexneri virulence. Mol Microbiol 23(4):765–775.