Abstract
By modulating the cellular redox state, the plasma membrane electron transport (PMET) is important in platelet biology; indeed, the oxidant/antioxidant balance plays a central role during activation of the coagulation pathway. None the less, in human platelets, the PMET system has not yet been fully characterized and the molecular identities of most components are unknown. Here, for the first time, the presence of the plasma membrane hydroquinone (NADH) oxidase Ecto-NOX1 in human platelets has been described. We found that Ecto-NOX1 expression is modulated by capsaicin: Indeed, it is positively regulated through a mechanism requiring binding of capsaicin to its receptor, namely the transient receptor potential vanilloid subtype 1 (TRPV1). Ligand-receptor interaction triggers a signalling cascade leading to ROS production, which in turn enhances expression and activity of Ecto-NOX1. Redox regulation of Ecto-NOX1 may be important to platelet recruitment and activation during inflammatory diseases.
| List of abbreviations: | ||
| CM-H2DCFDA, | = | 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester |
| DTNB, | = | 5-5′dithiobis(2-nitrobenzoic acid) |
| 5′-IRTX, | = | 5′-iodo-resiniferatoxin |
| mPMS, | = | 1-methoxy-5-methyl-phenazinium methyl sulphate |
| NAC, | = | N-acetyl-L-cysteine |
| PMET, | = | plasma membrane electron transport |
| TRPV1, | = | transient receptor potential vanilloid subtype 1 |
| WST-1, | = | 2-(4-iodophenyl)-3-(4-nitrophenyI)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt |
Introduction
In all living cells, the biological function of the plasma membrane electron transport (PMET) system is to maintain the redox balance inside and outside the cell. It is well known that this system transfers intracellular electrons (derived from NADH, NADPH or ascorbic acid) (Berridge and Tan Citation2000, VanDuijn et al. Citation2001) to extracellular acceptors, including oxygen and ascorbyl free radical (VanDuijn et al. Citation2001, Herst et al. Citation2004, Herst and Berridge Citation2007), through the activity of several enzymes located in the plasma membrane (Ly and Lawen Citation2003).
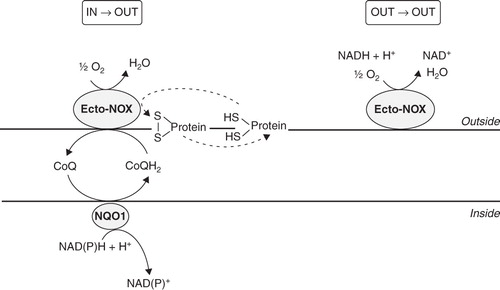
The PMET-mediated modulation of the cellular redox state is important in platelet biology; indeed, oxidant/antioxidant balance plays a central role during activation of the coagulation pathway (Krötz et al. Citation2004). None the less, in human platelets the PMET system has not yet been fully characterized and the molecular identities of most components are unknown (Del Principe et al. Citation2009). To date, the best characterized components of the platelet PMET system are the NAD(P)H oxidase (NOX2) (Seno et al. Citation2001, Krötz et al. Citation2002, Stokes et al. Citation2007) and some thiol-related enzymes (protein disulphide isomerase, endoplasmic reticulum protein 5 and glutathione reductase) (Essex et al. Citation1995, Citation2004, Jordan et al. Citation2005). NOX2 is a member of the NOX family, first identified in phagocytic cells, which catalyzes the one-electron transfer from NADPH to molecular oxygen, thus generating superoxide anions (O2.-). Platelets are not phagocytic cells, therefore platelet NOX2 may play a different role, principally related to activation and recruitment (Begonja et al. Citation2005). Thiol-related enzymes are involved in the rearrangement of membrane disulfide bonds important to platelet aggregation, secretion and post-aggregation events. Platelets also have a membrane NADH oxidase activity, not yet unequivocally ascribed to a specific protein; however, some features, such as cyanide insensitivity, NADH preference and periodicity, are shared with external NADH oxidases, also called ‘Ecto-NOXs’ (Del Principe et al. Citation1977, Peter et al. Citation2000). These enzymes are particular, as they are not trans-membrane proteins, but instead they are localized on the external surface of eukaryotic cells (Morré Citation1998, Yantiri and Morré Citation2001). Ecto-NOX proteins show hydroquinone (NADH) oxidase and protein disulfide–thiol interchange activities, cycling every 22–26 min, and in this way they modulate intra- and extra-cellular redox homeostasis, cell growth and survival (Morré and Morré Citation2003). Based on catalytic characteristics, three related, but distinct, Ecto-NOX proteins have been reported: A constitutive protein (cNOX or Ecto-NOX1), widely distributed among animals, plants and yeast, a tumour-associated protein, designated tNOX or Ecto-NOX2, and an aging-related protein (arNOX) present only in aged individuals (Morré et al. Citation2003, Jiang et al. Citation2008). Being located on the outer side of the plasma membrane, Ecto-NOXs allow electron transport from cytosolic NAD(P)H to acceptors at the cell surface, thus functioning as terminal oxidases of the PMET system (Kishi et al. Citation1999). It is noteworthy that pyridine nucleotides have recently been found in the extracellular compartment (Billington et al. Citation2006). Therefore, an alternative electron flow may exist: both donors and acceptors can be localized extracellularly, thus allowing an external (out-out) electron movement ().
Figure 1. Diagram of electron flows generated by Ecto-NOX activity. Electrons can move from inside to outside (in→out) or can be retained in the extracellular space (out→out). The in→out movement is directed from the intracellular NAD(P)H to the inter-membrane quinone (CoQ) and finally to extracellular electron acceptors, such as O2 or protein disulfides. In the out→out movement both the electron donor and acceptor are located outside the cell. NQO1, NAD(P)H:ubiquinone oxidoreductase. Ecto-NOX, hydroquinone (NADH) oxidase.
In this study we sought to identify the protein(s) responsible for platelet NADH oxidase activity. Here, for the first time, we show that platelets express Ecto-NOX1 protein, whose expression is up-modulated by capsaicin (8-methyl-N-vanilly-6-nonenamide), a typical vanilloid known to modulate the PMET system (Morré et al. Citation1995, Vaillant et al. Citation1996, Scarlett et al. Citation2005, Joung et al. Citation2007, Takahashi et al. Citation2008). Ecto-NOX1 up-modulation is achieved through capsaicin binding to the transient receptor potential vanilloid subtype 1 (TRPV1) and subsequent production of reactive oxygen species (ROS).
Materials and methods
Reagents
2-(4-iodophenyl)-3-(4-nitrophenyI)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) was obtained from Cayman Chemical Company (Ann Arbor, Michigan, USA). 5′-iodo-resiniferatoxin (5′-IRTX) was obtained from Tocris (Bristol, UK). All the other reagents, unless otherwise indicated, were from Sigma Chemical (St Louis, MO, USA).
Platelet isolation and drug treatments
After informed consent, platelets were isolated from blood samples obtained from 12 healthy donors (20–40 years old) free of medication for at least the preceding 30 days. Blood was collected in plastic tubes and diluted 1:5 with ACD buffer (80 mM glucose, 25 mM citric acid 45 mM sodium citrate). The platelet pellet was prepared by centrifuging plasma through a Ficoll layer (23% w/v) at 80 g for 20 min, as previously described (Savini et al. Citation2007); after washings, platelets were resuspended in tyrode's buffer (100 mM HEPES, 1.3 M NaCl, 29 mM KCl, 10 mM NaHCO3).
Capsaicin treatments were performed by adding capsaicin dissolved in ethanol just before use; 100 μM capsaicin corresponded to 0.004% ethanol final concentration. The time length of capsaicin treatment was 45 min in all experiments, except for time course experiments. After incubation, cells were rinsed with tyrode's buffer (in order to remove capsaicin) and collected to perform subsequent analyses. Where indicated, platelets were pre-treated for 10 min either with 50 μM capsazepine or 20 μM 5′-IRTX, or for 15 min with 5 mM N-acetyl-L-cysteine (NAC), before incubation with capsaicin.
Western blot
After treatments, platelets were resuspended in lysis buffer (25 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5% SDS) and disrupted by three freeze-thawing cycles. Plasma membranes were separated on a sucrose step gradient, as already reported (Dean and Quinton Citation1995). Membrane fractions (10 μg) were subjected to SDS-PAGE on a 10% polyacrylamide gel and then electroblotted onto a PVDF membrane. Blots were blocked with 5% non fat dry milk (Biorad, Hercules, CA, USA) and then incubated with anti-TRPV1 (sc-12498), anti-tubulin (sc-9104) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-Ecto-NOX1 (H00055068-B01 and H00055068-B01P; Abnova, Taipei City, Taiwan) primary antibodies. After washings and incubation with the horseradish peroxidase-conjugated secondary antibody, detection was carried out with ECL (Amersham, Arlington Heights, IL, USA).
Competition experiments were carried out by incubating the anti-TRPV1antibody solution with a 100-fold molar excess of TRPV1 blocking peptide (sc-12498P) for 3 h at room temperature, before performing Western blot analysis.
NAD(P)H oxidase activity
NADH and NADPH oxidase activities were determined by monitoring the decrease in absorbance (340 nm) of exogenously added nucleotides (Morré, Citation2004). Briefly, intact platelets (1 × 108) were incubated in 1 ml of tyrode's buffer containing 0.2 mM NADH or 0.2 mM NADPH at 37°C with continuous stirring, and rates of NADH or NADPH oxidation were calculated using the extinction coefficient 6.2 mM-1 cm-1. NAD(P)H oxidation was also monitored in purified plasma membrane preparations (100–200 μg of proteins) with slight modifications: in particular, 1 mM KCN was added to the mixture in order to inhibit any potential contaminating mitochondrial oxidase activity. In some experiments, NADH oxidation was monitored after heating to 50°C for 10 min and removing heat-denatured proteins by centrifugation at 6000 rpm for 5 min (Yantiri and Morré Citation2001).
NAD(P)H oxidase activity was also measured by monitoring the reduction of WST-1, a cell-impermeable artificial electron acceptor (Geng et al. Citation2009). Briefly, intact platelets (1 × 108) were incubated with 0.5 mM WST-1 and 0.2 mM NADH or 0.2 mM NADPH. All experiments were performed at 37°C, with continuous stirring, and WST-1 formazan production was monitored in real time by the increase of absorption at 450 nm. Rates of WST-1 reduction were expressed as nmol WST-1 reduced/min/mg protein (extinction coefficient = 37 mM−1cm−1).
In some experiments, enzymatic activity measurements have been performed in the presence of 100 μM capsaicin, by directly adding the drug to the assay mixture.
Trans-PMET activity
Trans-PMET activity was evaluated by measuring WST-1-reduction in the presence of 1-methoxy-5-methyl-phenazinium methyl sulphate (mPMS), which is a stable electron-transport mediator between NAD(P)H and WST-1 (Hisada and Yagi Citation1977). Washed platelets (1 × 109) were incubated in 1 ml of tyrode's buffer containing 0.5 mM WST-1 and 20 μM mPMS, and WST-1 reduction was monitored in real time at 450 nm. In some experiments, enzymatic activity measurements have been performed in the presence of 100 μM capsaicin, by directly adding the drug to the assay mixture.
Intracellular ROS
Intracellular ROS generation was measured by using the fluorescent probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Molecular Probes Inc., Eugene, OR, USA). After treatment, platelets were incubated with 10 μM CM-H2DCFDA for 20 min at 37°C in the dark. Radical formation was assessed by flow cytometry in a FACSCalibur Flow Cytometer (Becton Dickinson, CA, USA). CM-H2DCFDA mean fluorescence was registered at 530 nm (bandwidth 30 nm), exciting at 488 nm using a 15 mW Argon laser. One hundred thousand events were evaluated for each analysis.
Sulfhydryl groups and glutathione content
Surface sulfhydryl groups were evaluated by using the membrane-impermeant reagent 5-5′dithiobis(2-nitrobenzoic acid) (DTNB). Briefly, 1 × 109 platelets in 1 ml tyrode's buffer were incubated with 0.2 mM DTNB for 15 min at 37°C. Platelets were pelleted (12,000 rpm for 5 min) and 2-nitro-5-thiobenzoic acid was measured in the supernatant at 412 nm (extinction coefficient = 14.150 mM-1 cm-1). Total sulfhydryl groups were evaluated in lysed platelets by the same assay, and the intracellular amounts were obtained by difference (Savini et al. Citation2007). Intracellular glutathione content was quantified by a DTNB-glutathione reductase recycling assay as described (Anderson Citation1985).
Statistical analysis
Statistical analysis was conducted with the program GraphPad Prism 4.00 (GraphPad Software Inc., La Jolla, CA, USA). Values are presented as the means ± SE. Either paired t-test or analysis of variance (ANOVA) followed by Bonferroni post hoc comparisons were performed to evaluate differences between samples. Significant differences were accepted at p < 0.05.
Results
Ecto-NOX1 is present in human platelets
We first investigated the NAD(P)H oxidase system, by evaluating cofactor specificity (NADH or NADPH) and membrane localization of the enzymes. We evaluated trans-membrane activity by monitoring the reduction of the cell-impermeable tetrazolium salt WST-1, as a result of intracellular electrons flowing outwards via the intermediate electron acceptor mPMS. We also assessed surface activity either by following oxidation of exogenously added NADH or NADPH or by measuring the WST-1 reduction in the presence of exogenous pyridine nucleotides. Platelets were unable to reduce WST-1, unless intermediate electron acceptor or exogenous electron donor were added to the assay mixture. WST-1 reduction in the presence of mPMS was 0.12 ± 0.01 nmol/min/mg of protein, while it was 2.1 ± 0.2 nmol/min/mg of protein, in the presence of exogenous NADH. The ratio between these two activities was much higher than that reported by Berridge and Tan (Citation2000) for other cell types, indicating that platelets possess an efficient surface oxidase activity. Platelets also showed an electron donor preference, since the rate of NADH-dependent WST-1 reduction was significantly higher than that of NADPH-dependent WST1 reduction (). It has been reported that the Ecto-NOX proteins may be responsible for cell surface reduction of WST-1 (Geng et al. Citation2009); none the less, this tetrazolium salt has also been used for detection of superoxide or superoxide-generating enzymes (Oritani et al. Citation2004, Tan and Berridge Citation2010). In order to circumvent the misunderstanding that could come from the use of WST-1, we also monitored the disappearance of NAD(P)H at 340 nm. Similar results were obtained, in that oxidation of exogenous NADH appeared to be more efficient than NADPH oxidation (). Experiments carried out on purified plasma membrane preparations proved that this activity was cyanide-insensitive and heat-resistant, as it was fully retained after incubation at 50°C for 10 min (data not shown); these biochemical features support the presence of one or more Ecto-NOX enzymes on platelet plasma membranes (Kishi et al. Citation1999, Morré, Citation2004). In addition, the finding that most of the NADH oxidase activity (80 ± 7%; n = 12, p < 0.05) was capsaicin-insensitive, allowed us to hypothesize the presence of Ecto-NOX1 (Jiang et al. Citation2008). Indeed, Ecto-NOX2 activity has been reported to be largely affected by capsaicin (Yantiri and Morré Citation2001) and its expression is restricted to tumour cells (Morré Citation2004). Also arNOX is unlikely the candidate, as our samples derived from individuals aged below 40 years (Morré et al. Citation2003).
Figure 2. Cofactor specificity and identity of the platelet NAD(P)H oxidase system. (A) NAD(P)H oxidase activity and NAD(P)H-dependent WST-1 reduction. Enzyme activities were measured spectrophotometrically, as reported under Materials and methods. Values are the means of five independent experiments, each performed in triplicate. *p < 0.001 vs. NADH-containing assays. (B) Western blot analysis of Ecto-NOX1 protein. Platelet plasma membranes (PLT) were immunoblotted with anti-Ecto-NOX1 antibody. Positive control is represented by cellular extracts from lymphocytes (Lympho). The arrow points to Ecto-NOX1 band; molecular weight standards are shown on the right. The radiograph is representative of four similar experiments.
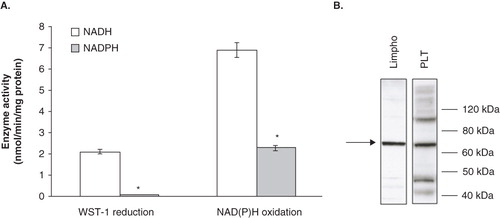
To further support our hypothesis, we performed Western blot analysis on membrane fractions. In agreement with biochemical results, the Ecto-NOX1 antibody recognized a protein band of about 70 kDa (), which corresponds to the molecular weight reported for full length Ecto-NOX1 protein (Jiang et al. Citation2008).
Finally, the Ecto-NOX1 antibody markedly inhibited the NADH oxidation (75% inhibition), thus demonstrating that Ecto-NOX1 appeared to be the main enzyme responsible for plasma membrane oxidase activity.
Ecto-NOX1 expression may be modulated by perturbations in the PMET system
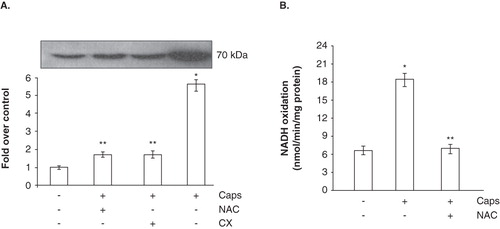
Next, we investigated if perturbations in the PMET system could alter Ecto-NOX1 expression. To this end, we incubated platelets in capsaicin-containing buffer, as this typical vanilloid has been shown to modulate the PMET system (Morré et al. Citation1995, Vaillant et al. Citation1996, Scarlett et al. Citation2005, Joung et al. Citation2007, Takahashi et al. Citation2008). After incubation and washings (in order to remove capsaicin), Western blot analysis and enzymatic activity were performed. The compound induced Ecto-NOX1 expression and cycloheximide experiments suggested that Ecto-NOX1 could be modulated at the translational level (). Interestingly, we found that NAC counteracted the effects of capsaicin, thus suggesting an involvement of ROS in the capsaicin-induced signalling leading to enhanced Ecto-NOX1 expression (). The raise in Ecto-NOX1 expression paralleled the increase in NADH oxidase activity: indeed, NADH oxidation significantly increased in capsaicin-treated platelets, in a NAC-sensitive manner ().
Figure 3. NADH-dependent WST-1 reduction and expression of Ecto-NOX1. (A) Western blot analysis of Ecto-NOX1. Platelets were left untreated or treated with 100 μM capsaicin (Caps) for 45 min, and immunoblotted with anti-Ecto-NOX1 antibody. The second bar represents platelets pre-treated with NAC for 15 min, before incubation with capsaicin; the third bar represents platelets incubated with capsaicin in the presence of 1 mM cycloheximide (CX). The radiograph is representative of four similar experiments. The histogram represents the densitometric analysis of autoradiography. Values are the means ± SE of three independent experiments, values are reported as fold over control, arbitrarily set to 1, after normalization with tubulin. *p < 0.001 vs. untreated platelets; **p < 0.05 vs. capsaicin. (B) NADH oxidation. Platelets were treated as in A and NADH oxidase activity was detected in whole cells, as described in Materials and methods.
Capsaicin regulates Ecto-NOX1 expression through vanilloid receptor and ROS
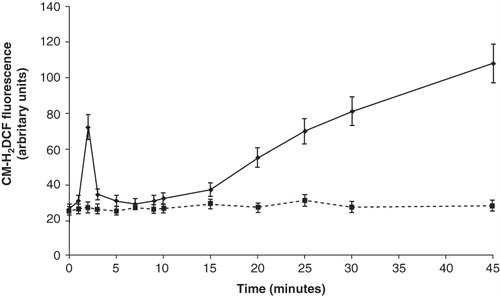
Based on data obtained with NAC, we then assessed if capsaicin induced changes in redox state. Time course experiments () showed that ROS levels, apart from a brief spike at 2 min, began to rise after 15 min of incubation with capsaicin, reaching a 4-fold increase, if compared with basal levels. The capsaicin-evoked ROS increase was completely annulled by NAC (data not shown).
Figure 4. Time course experiment of ROS production by capsaicin. Platelets were incubated with 100 μM capsaicin for the indicated times, before assessing radical formation by flow cytometry. Values are the means ± SE of five independent experiments. Dashed line: control platelets. Solid line: capsaicin-treated platelets.
Capsaicin also caused a 30% decrease in total glutathione content and a 2-fold increase in the GSSG/GSH ratio, as well as a 40% decrease in the levels of ascorbic acid (). We also found that capsaicin reduced both surface and intracellular sulfhydryl groups, although the effects were significant only on the former ().
Table I. Capsaicin modifies platelet redox state.
From these data, we concluded that capsaicin induced ROS elevation, thus promoting oxidative stress. In the beginning, platelets scavenged these reactive species, as they possessed high concentrations of antioxidant molecules (including glutathione and ascorbic acid), thus explaining the return of ROS levels to basal values within the first minutes. None the less, ROS production led to depletion in these antioxidant molecules, thus compromising the scavenging ability of platelets; as a result, ROS levels continuously increase (see ).
As capsaicin could bind to the TRPV1 receptor, recently shown to be expressed in human platelets (Harper et al. Citation2009), we investigated whether the drug effects on the redox state were exerted through a vanilloid receptor-dependent pathway. First, we investigated the presence of TRPV1 protein in platelet samples, as our purification protocol differed from that adopted by Harper et al (Citation2009). Western blot analysis showed two reactive bands with an apparent molecular weight of about 70 and 100 kDa: only the immunoreactive band with the highest molecular weight corresponded to the TRPV1 receptor, as confirmed by competition studies performed with the blocking peptide (). To investigate the involvement of TRPV1 in capsaicin-induced ROS production, an ultra potent agonist (resiniferatoxin) (Szolcsanyi et al. Citation1990) and two specific antagonists (5′-IRTX and capsazepine) (Dickenson and Dray Citation1991, Wahl et al. Citation2001) of the vanilloid receptor were used. showed that activation of the TRPV1 receptor by either capsaicin or resiniferatoxin resulted in a strong induction of ROS generation. Accordingly, receptor antagonists exerted the opposite effects; in fact, the 4-fold increase elicited by capsaicin alone was halved when platelets were pre-treated with either 20 μM 5′-IRTX or 50 μM capsazepine.
Figure 5. TRPV1 involvement in Ecto-NOX1 modulation. (A) Western blot analysis of TRPV1 receptor. Platelet extracts were immunoblotted with anti-TRPV1 antibody, alone or with TRPV1-specific blocking peptide. The radiographs are representative of four similar experiments. (B) ROS production with TRPV1 agonists and antagonists. Platelets were left untreated or treated with capsaicin (Caps, 100 μM) and resiniferatoxin (RTX, 10 μM) for 45 min; some samples were also pre-treated either with capsazepine (Cpz, 50 μM) or 5′-iodo-resiniferatoxin (5′-IRTX, 20 μM) for 10 min, before incubation with capsaicin. Data are the means ± SE of three independent experiments, each performed in triplicate. *p < 0.001 vs. untreated platelets; **p < 0.05 vs. capsaicin. (C) Western blot analysis of Ecto-NOX1 with TRPV1 agonists and antagonists. Platelets were left untreated or treated with 100 μM capsaicin for 45 min; some samples were also pre-treated with capsazepine for 10 minutes, before incubation with capsaicin. The radiograph is representative of four similar experiments. The histogram represents the densitometric analysis of autoradiography. Values are the means ± SE of three independent experiments, values are reported as fold over control, arbitrarily set to 1, after normalization with tubulin. *p < 0.001 vs. untreated platelets; **p < 0.05 vs. capsaicin.
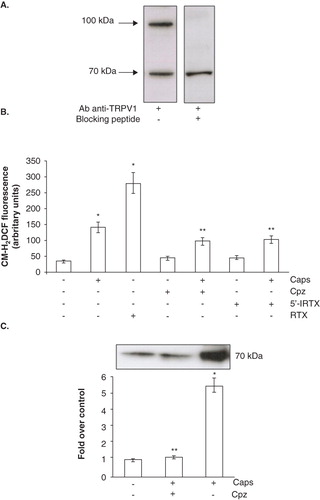
Finally, we investigated if TRPV1-mediated ROS production could be responsible for Ecto-NOX1 up-modulation. To this end, we pre-incubated platelets with capsazepine, before capsaicin treatment. We found that this TRPV1 antagonist efficiently prevented the capsaicin-triggered increase of Ecto-NOX1 protein (), thus confirming our hypothesis.
Discussion
The PMET system is crucial for blood coagulation and thrombosis as ROS and thiol changes, generated by several plasma membrane enzymes, modulate activation of platelets, as well as their interaction with leukocytes (Avigliano et al. Citation2008, Del Principe et al. Citation2009). The basic components of the PMET system found on platelet surface are not yet fully characterized. Although some elements have already been identified (such as the phox-NOX proteins) (Seno et al. Citation2001), none the less a great deal remains to be done; indeed, molecular data are lacking and the available information refers only to enzymatic activities measured with different experimental approaches. Now, for the first time, we give evidence for the presence of Ecto-NOX1 in human platelets, whose expression may be translationally modulated by capsaicin-evoked signalling.
Our data show that resting platelets have both trans-membrane and surface enzymatic activities. Our findings put forward Ecto-NOX1 as the main surface oxidase. Firstly, the observed activity preferentially oxidizes NADH and is localized on platelet membranes, as proved by NADH oxidation measurements performed in intact cells and purified membranes, as well as by Western blot analysis; secondly, the activity is cyanide-insensitive and heat-resistant; finally, the Ecto-NOX1 antibody markedly inhibits this activity.
In contrast to Ecto-NOX2, Ecto-NOX1 has been demonstrated to be capsaicin-insensitive (Jiang et al. Citation2008). None the less, we report that this drug triggers a signalling cascade able to translationally modulate Ecto-NOX1 expression; the up-regulation appears to be achieved via activation of TRPV1 (the natural receptor for capsaicin) and the generation of ROS. It should be recalled that TRPV1 activation may lead to biphasic ROS production. In capsaicin-treated platelets, we found a spike in ROS concentration as early as 2 min followed by a gradual rise after 15 min. Thus, ligand-receptor binding activates the signalling cascade responsible for the early events (such as intracellular Ca2+ increase) as already reported (Harper et al. Citation2009), but the TRPV1-dependent Ecto-NOX1 activation appears to rely on prolonged activation of the receptor, since it is a later event.
From these data, a more detailed view of the platelet PMET system can be provided; in particular, the presence of Ecto-NOX1 and the possibility that it is a redox-sensitive protein may be of importance in terms of electron movement during platelet activation and cell-to-cell interactions (platelet-platelet, platelet-leukocyte, platelet-endothelial cells) (Hidalgo et al. Citation2009, Looney and Matthay Citation2009). Tissue injury usually promotes the release of several mediators, including histamine, anandamide, prostaglandins and cytokines, which directly or indirectly activate TRPV1 (Khairatkar-Joshi and Szallasi Citation2009). Thus, one can speculate that activators of this receptor may promote ROS increase, which in turn enhances Ecto-NOX1 expression. In addition, the concentration of purine nucleotides in extracellular body fluids, usually present in the sub-micromolar range (Billington et al. Citation2006), can increase during inflammation, thus enhancing Ecto-NOX activity. In a wider point of view, exposure of platelets to low-level oxidative stress or growth factors, triggering ROS increase, may result in upregulation of the plasma membrane-bound NADH oxidase.
In conclusion, the potential redox sensitivity of Ecto-NOX1 may be a novel field of interest, especially keeping in mind that platelet activation and aggregation strictly depend on redox state. To this purpose, it should be recalled that Ecto-NOX1 has a disulfide isomerase activity, which may be important for regulating the oxidative state of sulphydryl groups present in key proteins involved in platelet functions. It should be interesting to further dissect this problem in the future, in order to establish the physiological role of Ecto-NOX1 in platelet biology.
Acknowledgements
We are grateful to Prof. G. Mignogna and Dr A. Giorgi for assistance and useful discussion.
Declaration of interest: This work was supported by a grant from the Italian Ministry MIUR (Ministero dell'Istruzione, dell'Università e della Ricerca). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Anderson ME. 1985. Determination of glutathione and glutathione disulfide in biological samples, Methods Enzymol 113:548–555.
- Avigliano L, Savini I, Catani MV, Del Principe D. 2008. Trans-plasma membrane electron transport in human blood platelets. Mini Rev Med Chem 8:555–563.
- Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, Walter U. 2005. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 106:2757–2760.
- Berridge MV, Tan AS. 2000. High-capacity redox control at the plasma membrane of mammalian cells: Trans-membrane, cell surface, and serum NADH-oxidases. Antioxid Redox Signal 2:231–242.
- Billington RA, Bruzzone S, De Flora A, Genazzani AA, Koch-Nolte F, Ziegler M, Zocchi E. 2006. Emerging functions of extracellular pyridine nucleotides. Mol Med 12:324–327.
- Dean WL, Quinton TM. 1995. Distribution of plasma membrane Ca2+-ATPase and inositol 1,4,5trisphosphate receptor in human platelet membranes. Cell Calcium 17:65–70.
- Del Principe D, Frega G, Savini I, Catani MV, Rossi A, Avigliano L. 2009. The plasma membrane redox system in human platelet functions and platelet-leukocyte interactions. Thromb Haemost 101:284–289.
- Del Principe D, Menichelli A, Damiano AM, Coppola L, Sabetta G. 1977. Cyanide insensitive oxidase in platelets of newborn infant. Thromb Haemost 37:339–343.
- Dickenson AH, Dray A. 1991. Selective antagonism of capsaicin by capsazepine: Evidence for a spinal receptor site in capsaicin-induced antinociception. Br J Pharmacol 104:1045–1049.
- Essex DW, Chen K, Swiatkowska M. 1995. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood 86:2168–2173.
- Essex DW, Li M, Feinman RD, Miller A. 2004. Platelet surface glutathione reductase-like activity. Blood 104:1383–1385.
- Geng L, Rachakonda G, Morré DJ, Morré DM, Crooks PA, Sonar VN, Roti JL, Rogers BE, Greco S, Ye F, Salleng KJ, Sasi S, Freeman ML, Sekhar KR. 2009. Indolyl-quinuclidinols inhibit ENOX activity and endothelial cell morphogenesis while enhancing radiation-mediated control of tumor vasculature. FASEB J 23:2986–2995.
- Harper AG, Brownlow SL, Sage SO. 2009. A role for TRPV1 in agonist-evoked activation of human platelets. J Thromb Haemost 7:330–338.
- Herst PM, Berridge MV. 2007. Cell surface oxygen consumption: A major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim Biophys Acta 1767:170–177.
- Herst PM, Tan AS, Scarlett DJ, Berridge MV. 2004. Cell surface oxygen consumption by mitochondrial gene knockout cells. Biochim Biophys Acta 1656:79–87.
- Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. 2009. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 15:384–391.
- Hisada R, Yagi T. 1977. 1-methoxy-5-methylphenazinium methyl sulphate, a photochemically stable electron mediator between NADH and various electron acceptors. J Biochem 82:1469–1473.
- Jiang Z, Gorenstein NM, Morré DM, Morré DJ. 2008. Molecular cloning and characterization of a candidate human growth-related and time-keeping constitutive cell surface hydroquinone (NADH) oxidase. Biochemistry 47:14028–14038.
- Jordan PA, Stevens JM, Hubbard GP, Barrett NE, Sage T, Authi KS, Gibbins JM. 2005. A role for the thiol isomerase protein ERP5 in platelet function. Blood 105:1500–1507.
- Joung EJ, Li MH, Lee HG, Somparn N, Jung YS, Na HK, Kim SH, Cha YN, Surh YJ. 2007. Capsaicin induces heme oxygenase-1 expression in HepG2 cells via activation of PI3K-Nrf2 signaling: NAD(P)H:quinone oxidoreductase as a potential target. Antioxid Redox Signal 9:2087–2098.
- Khairatkar-Joshi N, Szallasi A. 2009. TRPV1 antagonists: The challenges for therapeutic targeting. Trends Mol Med 15:14–22.
- Kishi T, Morré DM, Morré DJ. 1999. The plasma membrane NADH oxidase of HeLa cells has hydroquinone oxidase activity. Biochim Biophys Acta 1412:66–77.
- Krötz F, Sohn HY, Gloe T, Zahler S, Riexinger T, Schiele TM, Becker BF, Theisen K, Klauss V, Pohl U. 2002. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 100:917–924.
- Krötz F, Sohn HY, Pohl U. 2004. Reactive oxygen species: Players in the platelet game. Arterioscler Thromb Vasc Biol 11:1988–1996.
- Looney MR, Matthay MA. 2009. Neutrophil sandwiches injure the microcirculation. Nat Med 15:364–366.
- Ly JD, Lawen A. 2003. Transplasma membrane electron transport: Enzymes involved and biological function. Redox Rep 8:3–21.
- Morré DJ, Chueh PJ, Morré DM. 1995. Capsaicin inhibits preferentially the NADH oxidase and growth of transformed cells in culture. Proc Natl Acad Sci USA 92:1831–1835.
- Morré DJ, Morré DM. 2003. Cell surface NADH oxidases (ECTO-NOX proteins) with roles in cancer, cellular time-keeping, growth, aging and neurodegenerative diseases. Free Radic Res 37:795–808.
- Morré DJ. 1998. NADH oxidase: A multifunctional ectoprotein of the eukaryotic cell surface. In: Asard H, Bérczi A, Caubergs R, editors. Plasma membrane redox systems and their role in biological stress and disease. Dordrecht: Kluwer Academic Publishers. pp 121-156.
- Morré DJ. 2004. Quinone oxidoreductases of the plasma membrane. Methods Enzymol 378:179–199.
- Morré DM, Guo F, Morré DJ. 2003. An aging-related cell surface NADH oxidase (arNOX) generates superoxide and is inhibited by coenzyme Q. Mol Cell Biochem 254:101–109.
- Oritani T, Fukuhara N, Okajima T, Kitamura F, Ohsaka T. 2004. Electrochemical and spectroscopic studies on electron-transfer reaction between novel water-soluble tetrazolium salts and a superoxide ion. Inorganica Chimica Acta 357:436–442.
- Peter AD, Morrè DJ, Morrè DM. 2000. A light-responsive and periodic NADH oxidase activity of the cell surface of tetrahymena and of human buffy coat cells. Antioxid Redox Signal 2:289–300.
- Savini I, Catani MV, Arnone R, Rossi A, Frega G, Del Principe D, Avigliano L. 2007. Translational control of the ascorbic acid transporter SVCT2 in human platelets. Free Radic Biol Med 42:608–616.
- Scarlett DJ, Herst PM, Berridge MV. 2005. Multiple proteins with single activities or a single protein with multiple activities: The conundrum of cell surface NADH oxidoreductases. Biochim Biophys Acta 1708:108–119.
- Seno T, Inoue N, Gao D, Okuda M, Sumi Y, Matsui K, Yamada S, Hirata KI, Kawashima S, Tawa R, Imajoh-Ohmi S, Sakurai H, Okoyama M. 2001. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb Res 103:399–409.
- Stokes KY, Russell JM, Jennings MH, Alexander JS, Granger DN. 2007. Platelet-associated NAD(P)H oxidase contributes to the thrombogenic phenotype induced by hypercholesterolemia. Free Radic Biol Med 43:22–30.
- Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM. 1990. Resiniferatoxin: An ultrapotent selective modulator of capsaicin-sensitive primary afferent neurons. J Pharmacol Exp Ther 255:923–928.
- Takahashi T, Okuno M, Tadashi O, Kishi T. 2008. NADPH-dependent coenzyme Q reductase is the main enzyme responsible for the reduction of non-mitochondrial CoQ in cells. BioFactors 32:59–70.
- Tan AS, Berridge MV. 2010. Evidence for NAD(P)H:quinone oxidoreductase 1 (NQO1)-mediated quinone-dependent redox cycling via plasma membrane electron transport: A sensitive cellular assay for NQO1. Free Radic Biol Med 48:421–429.
- Vaillant F, Larm JA, McMullen GL, Wolvetang EJ, Lawen A. 1996. Effectors of the mammalian plasma membrane NADH-oxidoreductase system Short-chain ubiquinone analogues as potent stimulators. J Bioenerg Biomembr 28:531–540.
- VanDuijn MM, Van der Zee J, Van den Broek PJ. 2001. The ascorbate-driven reduction of extracellular ascorbate free radical by the erythrocyte is an electrogenic process. FEBS Lett 491:67–70.
- Wahl P, Foged C, Tullin S, Thomsen C. 2001. Iodo-resiniferatoxin, a new potent vanilloid receptor antagonist. Mol Pharmacol 59:9–15.
- Yantiri F, Morré DJ. 2001. Isolation and characterization of a tumor-associated NADH oxidase (tNOX) from the HeLa cell surface. Arch Biochem Biophys 391:149–159.