Abstract
The endoplasmic reticulum (ER) is a highly organized and specialized organelle optimized for the production of proteins. It is comprised of a highly interconnected network of tubules that contain a large set of resident proteins dedicated to the maturation and processing of proteins that traverse the eukaryotic secretory pathway. As protein maturation is an imperfect process, frequently resulting in misfolding and/or the formation of aggregates, proteins are subjected to a series of evaluation processes within the ER. Proteins deemed native are sorted for anterograde trafficking, while immature or non-native proteins are initially retained in the ER in an attempt to rescue the aberrant products. Terminally misfolded substrates are eventually targeted for turnover through the ER-associated degradation or ERAD pathway to protect the cell from the release of a defective product. A clearer picture of the identity of the machinery involved in these quality control evaluation processes and their mechanisms of actions has emerged over the past decade.
Introduction
Ribosome-attached secretory pathway cargo are co-translationally targeted to the endoplasmic reticulum (ER) translocon Sec61 by the signal recognition particle or SRP. Nascent chains co-translationally emerge into the ER lumen through this ER membrane pore in a vectorial manner (N- to C-terminus). As protein maturation begins co-translational and translocationally, the vectorial nature of these processes helps to taper the ensemble of possible intermediates by starting maturation with shorter nascent chains. These early events, which are assisted by a group of factors localized in close proximity to the Sec61 entry site, include protein folding, processing and modification. The majority of the secretory pathway customers are modified by N-linked glycans (Apweiler et al. Citation1999), and these modifications are used extensively as maturation and quality control tags that assist with the sorting process. This review will focus on the machinery and mechanics for how these sorting decisions are mediated with the help of N-linked glycans and their related factors.
Getting started: Translocation and early maturation events
Proteins containing the consensus N-linked glycosylation site Asn-X-Ser/Thr are co-translationally modified with a pre-assembled 14-member carbohydrate comprised of three glucoses, nine mannoses and two N-acetylglucosamines (Glc3Man9GlcNAc2; ) through the action of the oligosaccharyl transferase (OST). The addition of these large bulky hydrophilic modifications greatly alters the fundamental properties of the protein. The enzymatic transfer of the glycan requires that the Ser or Thr, situated two residues C-terminal to the Asn attachment site, position their hydroxyl side chain to render the Asn amide group more nucleophilic to support the progression of the transfer reaction. Therefore, this reaction mechanism favors the transfer of glycans that are situated on flexible regions. These regions are frequently located on the aqueous exposed surface of the protein. This optimally positions glycans for their role as maturation and quality control tags to support the recruitment of protein folding and degradation factors.
Figure 1. Schematic representation of N-linked glycan. (A) Structure of N-linked glycan including glucoses (green), mannoses (blue) and N-acetylglucosamines (gray) are indicated by triangles, spheres and rectangles, respectively. The branches on the glycan (A, B and C) are designated. Carbohydrate linkages are indicated. (B) Proposed structures of the glycans serving as degradation signals in ERAD. The glucoses have been trimmed after the calnexin cycle. The outer most B-branch mannose is trimmed by ER mannosidase I/Mns1p. The α1,2 mannose on the C-branch has been proposed to be removed by EDEM1/Htm1p. Trimming of C-branch mannose residues generates exposed α1,6 mannose, which has been proposed to be a degradation signal for the receptors such as Yos9p and OS-9/XTP3-B.
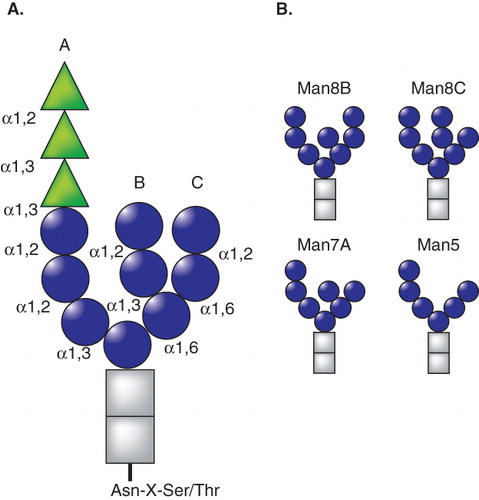
The composition of the carbohydrate tag is controlled by a series of glycosidases and glycosyltransferases that line the secretory pathway (). In the ER, glucosidase I initially removes terminal glucose residues creating diglucosylated (Glc2Man9GlcNAc2) side chains, which have recently been shown to support the recruitment of the ER-resident protein malectin (Schallus et al. Citation2008). Malectin was identified as a protein immunoisolated in aquaporin-2 containing vesicles, suggestive of it possibly playing a role in its maturation (Barile et al. Citation2005). As glucosidase I cleavage occurs rapidly co-translationally (Chen et al. Citation1995), this implies that malectin acts early in the maturation process perhaps during translocation (). However, further studies are required to understand the spectrum of malectin substrates, the timing and role in its assistance in cargo maturation.
Table I. ER maturation and quality control factors. The mammalian components are listed with Saccharomyces cerevisia e homologues denoted in parenthesis.
Figure 2. Glucose-dependent early protein maturation. After the entry of polypeptides into the ER, glucosidase I trims the first glucose. Diglucosylated glycans mediate interactions with malectin and glucosidase II trims the second glucose. This monoglucosylated glycan is the target of lectin-chaperones, calnexin (CNX) and calreticulin (CRT). Glucosidase II removes the final glucose to generate an unglucosylated glycan. Mature polypeptides are sorted for anterograde trafficking, while still immature or non-native proteins are reglucosylated by UGT1 to support calnexin/calreticulin rebinding. Eventually, terminally misfolded proteins are targeted to ERAD pathway for destruction.
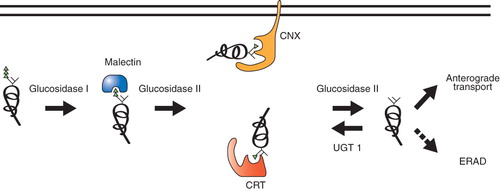
The second glucose is trimmed by glucosidase II to generate monoglucosylated glycans. These carbohydrates serve as attachment sites for the lectin chaperones calnexin, a type I membrane protein, and its soluble paralogue calreticulin (Ou et al. Citation1993, Peterson et al. Citation1995). The action of glucosidase II also occurs co-translationally supporting early co-translational binding to the lectin chaperones (Chen et al. Citation1995, Hebert et al. Citation1997, Deprez et al. Citation2005) (). Calnexin and calreticulin binding serves a number of roles. First, as both chaperones are ER retained through ER retention signals, binding to immature proteins keeps client proteins in the ER (Rajagopalan et al. Citation1994). Second, both chaperones are associated with an oxidoreductase ERp57, so binding recruits an oxidoreductase to the maturing chain that can assist in disulfide formation, reduction or rearrangement (Oliver et al. Citation1997, Zapun et al. Citation1998, Solda et al. Citation2006). Third, chaperone binding constrains or delays the folding of a protein in a region-specific manner; therefore, chaperone binding can direct the folding of a polypeptide by controlling the temporal accessibility of regions to fold (Daniels et al. Citation2003). Finally, the lectin chaperones protect vulnerable nascent chains from aggregation and degradation (Hebert et al. Citation1996, Vassilakos et al. Citation1996). Inhibition of glucose trimming using competitive inhibitors of glucosidases I and II such as castanospemine or deoxynojirimycin abolished calnexin and calreticulin binding and accelerated the turnover for a number of glycoproteins through the endoplasmic reticulum-associated degradation (ERAD) pathway (Kearse et al. Citation1994, Keller et al. Citation1998, Ayalon-Soffer et al. Citation1999, Liu et al. Citation1999, Chung et al. Citation2000, Molinari et al. Citation2002, Svedine et al. Citation2004). While stable cell lines lacking calnexin, calreticulin, or ERp57 have been established, the lectin chaperones and their associated oxidoreductase are essential for the survival of multicellular metazoans underscoring their importance in organismal homeostasis (Mesaeli et al. Citation1999, Denzel et al. Citation2002, Coe et al. Citation2010).
The cleavage of the final glucose by glucosidase II disrupts binding to the lectin chaperones (Hebert et al. Citation1995) (). Glucosidase II cleavage of the second and third glucoses is not processive as the substrate appears to require repositioning between cleavage events (Mackeen et al. Citation2009). Glucosidase II is comprised of α- and β-subunits. The α-subunit contains the catalytic activity, while the β-subunit possesses the ER retention signal as well as a Mannose-6 phosphate Receptor Homology (MRH) domain that influences substrate specificity (Deprez et al. Citation2005, Stigliano et al. Citation2009). The MRH domain helps in substrate recruitment and perhaps in its activity as the β-subunit appears to be critical for the accelerated cleavage of the final glucose to generate unglucosylated glycans (Deprez et al. Citation2005, Watanabe et al. Citation2009). As small proteins may be able to fold without the assistance of chaperones, it would be of interest to determine if glucosidase II trimming can be controlled to by-pass lectin chaperone binding so that these chaperones can focus their attention on more complex proteins that require their assistance for proper maturation. The number and location of glycans appears to dictate to some extent the molecular choreography or the chaperone binding profile for the individual chaperones (Hebert et al. Citation1997, Molinari and Helenius Citation2000, Wang et al. Citation2005, Citation2008). Molinari and Helenius (Citation2000) proposed the cotranslational model whereby prior interaction with BiP before the lectin chaperones only occurs if there is no glycan located within 50 amino acids of the N-terminus of the protein. While non-glycosylated proteins or regions may be aided by the Hsp70 chaperone BiP and protein disulfide isomerase family members such as PDI, glycans appear to provide the dominant chaperone signal. Together, the region-specific recruitment of the carbohydrate binding chaperones and the bulky hydrophilic nature of the glycan, likely inhibit Hsp70 binding as this family of chaperones recognizes hydrophobic patches in extended conformations (Blond-Elguindi et al. Citation1993). BiP can also serve as a back-up chaperone system in cases where lectin chaperone binding has been inhibited (Zhang et al. Citation1997).
The positioning of BiP at the translocon by the J-domain of Sec63 and its ability to directly bind polypeptides supports the early action of BiP in the protein maturation process. However, N-linked glycans route glycoproteins to the lectin chaperone assistance program. The lectin chaperone binding profile involves the initial binding to calnexin, the membrane integrated chaperone (Wang et al. Citation2005). In many cases, this is followed by association with calreticulin, if the substrate is soluble or if its glycans eventually extend deep into the lumen away from the membrane. The level of calnexin associated with the translocon complex can be regulated by the phosphorylation of its C-terminal cytoplasmic tail, which supports ribosome binding and translocon association (Chevet et al. Citation1999). The evolution of larger more complex proteins in multicellular organisms appears to have resulted in the development of an additional sophisticated chaperone system based on carbohydrate binding properties. A better understanding of the organization of the translocon-associated factors will aid in our knowledge of the maturation assembly line that appears to assist in the efficient maturation of secretory cargo.
UGT1 as an early quality control gatekeeper
Glucose trimming can support a single round of lectin chaperone binding, however rebinding to these chaperones or the so-called ‘calnexin cycle’ is controlled by the UDP-glucose:glycoprotein glucosyltransferase 1 (UGT1) (). UGT1 is a ∼ 170 kDa soluble ER resident protein comprised of two functional domains. The C-terminal domain provides the glucosyltransferase activity of the enzyme that uses the ER UDP-glucose pool as a source to transfer glucoses onto the A-branch of glycans attached to immature or non-native substrates ( and ). The majority of the enzyme is comprised of the N-terminal folding sensor domain for which the structure is unknown. Together, these activities are critical for its role as an important quality control gatekeeper that directs proteins for ER retention through rebinding to the lectin chaperones.
Biochemical studies using purified UGT1 have demonstrated that UGT1 recognizes surface-exposed hydrophobic regions on maturing proteins that are associated with non-native or unassembled proteins (Sousa and Parodi Citation1995, Trombetta and Helenius Citation2000, Keith et al. Citation2005). The analysis of glycopeptides reglucosylated by purified UGT1 discovered that regions immediately C-terminal to efficiently reglucosylated sites possessed oscillating extended regions of hydrophobicity (Taylor et al. Citation2003). Positioning of the hydrophobic region closer to synthetic glycans has also been shown to increase reglucosylation levels (Totani et al. Citation2009). These results suggest that UGT1 optimally acts on glycans proximal to hydrophobic structural perturbations.
In addition to retaining proteins in the ER, chaperone binding has also been proposed to perturb or delay the folding of proteins in a region-specific manner (Daniels et al. Citation2003, Wang et al. Citation2005, Pearse et al. Citation2008, Pearse and Hebert Citation2010). For multidomain proteins, it can be advantageous to control the order of domain folding. The non-sequential domain protein influenza hemagglutinin (HA) has an N-terminal segment that interacts with a C-terminal region, linked through a large loop disulfide bond. Efficient calnexin binding to the three glycans located on the N-terminal segment protects this domain from immature oxidation to form non-native disulfide linkages, and tethers it to the membrane until a medial domain folds and the C-terminal interacting region is translated (Daniels et al. Citation2003). The efficient reglucosylation of the N-terminal region of HA supports persistent chaperone binding, which helps to direct its efficient folding (Pearse et al. Citation2008). These results are suggestive of a larger role for UGT1 beyond targeting proteins for ER retention, which includes actively playing a role in directing the folding of large multidomain proteins by controlling region-specific chaperone binding.
UGT1 reglucosylation appears to be positioned within the ER to act at a late stage during ER maturation and quality control. Quantitative immunogold electron microscopy found UGT1 to be predominantly located in transitional ER/exit sites or pre-Golgi intermediates (Zuber et al. Citation2001). These results were consistent with more recent proteomics results that observed UGT1 levels to be the highest in smooth ER membrane preparations (Gilchrist et al. Citation2006). Finally, reglucosylation of HA was observed to solely occur post-translationally after release from the ribosome-translocon complex (Pearse et al. Citation2008). Together, these results are indicative of UGT1 playing a late role in the folding process. This supports the initial chaperone binding being directed by glucosidase II trimming prior to engaging the rebinding cycle or the reglucosylation activity that resides deeper into the ER away from the entry sites found in the rough ER.
There is growing amount of evidence indicating that reglucosylation by UGT1 and lectin chaperone rebinding can increase the maturation efficiency of a subset of glycoproteins as the maturation of a number of glycoproteins is significantly diminished in ugt1-/- cells (Molinari et al. Citation2005, Pearse et al. Citation2010). Furthermore, augmenting UGT1 reglucosylation activity by increasing UDP-glucose levels has been shown to enhance the maturation of transferrin (Wada et al. Citation1997). A link between UGT1 and the sorting of defective secretory cargo to the ERAD pathway is less certain. In vitro studies with purified proteins have demonstrated that UGT1 recognizes near native proteins (Caramelo et al. Citation2003, Citation2004), suggestive of it assisting proteins that are on pathway to reach their native state but perhaps just need a little more help or time to mature properly. In contrast, the overexpression of downstream ERAD factors such as EDEM1 can accelerate defective cargo extraction from the calnexin binding cycle and their subsequent degradation, suggesting that ERAD cargo may be caught in a futile chaperone binding cycle awaiting recognition for ERAD (Molinari et al. Citation2003, Oda et al. Citation2003). Additional studies will be required to determine if UGT1 is able to focus the efforts of the lectin chaperone binding cycle to proteins that are near native and can eventually be rescued to fold properly, or alternatively, if it can also help to retain defective cargo in the ER until recognition for ERAD sorting.
Mannose trimming for ERAD
Pharmacological and genetic analyses have revealed that the clearance of misfolded glycoproteins from the ER is linked to the processing of their N-linked glycans or more specifically mannose trimming. The involvement of mannose trimming for misfolded protein turnover was first reported for the heterologous expression of yeast prepro-α factor in rat pituitary cells as the inhibition of mannose trimming with deoxymannojirimycin, a competitive inhibitor of ER α-mannosidases, significantly stabilized prepro-αfactor (Su et al. Citation1993). These results were further verified in both yeast and mammalian cells using both inhibitors and the genetic deletion/knockdown of mannosidase activity (Jakob et al. Citation1998, Liu et al. Citation1999, Svedine et al. Citation2004, Bhamidipati et al. Citation2005, Avezov et al. Citation2008, Termine et al. Citation2009). An important caveat to note is that these approaches supported the global retention of mannose residues on all glycoproteins and was not limited to ERAD substrates. These findings led to the hypothesis that mannose trimming generated an ERAD signal that was recognized by downstream carbohydrate-binding proteins that aided in sorting aberrant proteins for degradation. Mannosidase activity was proposed to act slow; therefore, only glycoproteins that possessed non-native structures and had been retained in the ER would be trimmed of these particular mannose residues (Helenius Citation1994, Cabral et al. Citation2001). Initially, trimming was thought to only involve the removal of a single mannose from the B-arm to generate the so-called Man8B glycoforms (Man8GlcNAc2; ).
More recent results suggest that the putative ERAD carbohydrate signal involves more extensive trimming of mannose residues to glycoforms from Man7 to Man5 in which B and C branch mannoses are trimmed (). The involvement of truncated mannose residues in ERAD is supported by the analysis of the carbohydrate binding specificities for downstream quality control receptors, which will be discussed more thoroughly below. Importantly, when proteasome degradation is inhibited, ERAD substrates were retained in the ER with mannose residues trimmed to Man5-7 glycoforms (Ermonval et al. Citation2001, Frenkel et al. Citation2003, Hosokawa et al. Citation2003, Kitzmuller et al. Citation2003, Avezov et al. Citation2008). As inhibiting degradation resulted in the prolonged accumulation of the ERAD substrates in the ER, it cannot be ruled out that the block supported more extensive mannosidase access and trimming. Further complicating the identification of the ERAD signal, cytosolic free oligosaccharides apparently created by cytoplasmic endoglycosidase cleavage of retrotranslocated ERAD substrates accumulated as Man8B glycans in yeast (Hirayama et al. Citation2010). And in cells that transfer Man5GlcNAc2 glycans, mannosidase inhibitors still support the stabilization of ERAD substrates even though the glycans should not require further trimming for ERAD recognition (Ermonval et al. Citation2001).
As mannose deprived side chains appear to mark substrates for ERAD, the next question becomes what is the identity of the mannosidase(s) responsible for the remodeling of mannose arms of ERAD substrates? And how does this mannosidase distinguish non-native from native proteins? The ER possesses a small group of mannosidase-like proteins, which includes ER mannosidase I and the EDEM family of proteins. In budding yeast, these families are comprised of two proteins termed Mns1p (ER α-Mannosidase I orthologue) and Htm1p (the EDEM1 orthologue, also called Mnl1p) (Quan et al. Citation2008, Clerc et al. Citation2009). These two proteins appear to cooperate to support the extensive de-mannosylation of ERAD substrates. The initial trimming to Man8B by Mns1p created a substrate, which then could be further trimmed by Htm1p to generate the ERAD signal that involved the exposure of α1,6-bonded mannose residues of the Man7 side chains () (Clerc et al. Citation2009). Deletion of Mns1 supported ERAD substrate stabilization as the protein was no longer a substrate for the downstream mannosidase activity of Htm1p.
The trimming of mammalian ERAD substrates appears to be more complex as the ER glycosyl hydrolase family is expanded, and the characterization of the mannosidase activity using purified proteins has been enigmatic. The overexpression of ER α-Mannosidase I in cells facilitated the turnover of misfolded proteins, and resulted in the accumulation of Man8B, as well as more extensively trimmed glycans (Man7 or Man6) (Hosokawa et al. Citation2003). In vitro studies with ER α-Mannosidase I have found that at biological levels a single mannose is trimmed to create Man8B; however, at higher levels of the enzyme and 24 h of incubation additional α1,2-mannoses can be trimmed to produce Man5-7 glycans (Herscovics et al. Citation2002). As exogenously expressed ER α-Mannosidase I have been observed to accumulate in a perinuclear region upon proteasome inhibition, this has led to an alternative hypothesis that aberrant proteins are sorted to an ER quality control compartment where ER α-Mannosidase I is concentrated sufficiently to support more extensive trimming of the mannose residues on ERAD substrates (Avezov et al. Citation2008, Lederkremer Citation2009). However, obtaining elevated levels of ER α-Mannosidase I would appear to be problematic as it is not induced by the unfolded protein response (UPR) in yeast or mammals, and its turnover has been reported to be rapid (Travers et al. Citation2000, Hosokawa et al. Citation2001, Wu et al. Citation2007). Intriguingly, ER α-Mannosidase I has been found to be stabilized by EDEM1 suggesting that ER α-Mannosidase I levels may be tightly regulated (Termine et al. Citation2009).
Recent evidence suggests that the mammalian ERAD system might work in a similar manner to that proposed for the yeast system as EDEM family members may act as mannosidases situated downstream of the activity of ER α-Mannosidase I. EDEM1 was first discovered as a mammalian orthologue to the yeast Htm1p protein that contained a mannosidase-like domain but was believed to lack mannosidase activity (Hosokawa et al. Citation2001). The overexpression of EDEM1 accelerated the turnover of glycosylated ERAD substrates, whereas its knock down stabilized them (Molinari et al. Citation2003, Gnann et al. Citation2004, Gong et al. Citation2005). Elucidating the role of EDEM1 in the ERAD process has been problematic. While the overexpression of EDEM1 in cells has been found to be associated with the accelerated trimming of mannose residues, attempts to identify any mannosidase activity using purified proteins have failed (Olivari et al. Citation2006, Cormier et al. Citation2009, Mikami et al. Citation2010, Hosokawa et al. Citation2010b). This has led to a couple of contrasting models for the function of EDEM1 in ERAD.
In the first model, EDEM1 possesses specialized mannosidase activity that acts on aberrant proteins to aid in the generation of an ERAD signal. The overexpression of EDEM1 in cells was associated with increased mannose trimming as determined by an increase in mobility of glycosylated ERAD substrates by SDS-PAGE or the composition of glycans released from ERAD substrates as analyzed by HPLC (Olivari et al. Citation2006, Cormier et al. Citation2009, Hosokawa et al. Citation2010b). EDEM1 overexpression also enhanced A-branch mannose trimming in B3F7 cells, as this cell lines transfers Man5 glycans lacking B- and C-branch mannose residues (Olivari et al. Citation2006). In contrast, the overexpression of wild type but not mannosidase domain mutant forms of EDEM1 supported the trimming of C-branch mannose residue of alpha-1-antitrypsin null Hong Kong to accumulate Man8C glycans () (Hosokawa et al. Citation2010b). While the primary structure of EDEM1 and the analysis of glycoproteins after EDEM1 overexpression are consistent with EDEM1 possessing mannosidase activity, the demonstration of such an activity is lacking for studies using purified protein and conflicting results are available as to the nature of the trimmed species (see above) (Olivari et al. Citation2006, Mikami et al. Citation2010, Hosokawa et al. Citation2010b).
An alternative model proposes that EDEM1 uses it mannosidase-like domain to bind glycans as a quality control receptor that recognizes misfolded substrates and sorts them for degradation (Molinari et al. Citation2003, Oda et al. Citation2003, Wang and Hebert Citation2003). EDEM1 was identified as an ER stress-induced gene with homology to conserved residues of ER Mannosidase I active site, however, it lacked a couple of cysteine residues thought to be required for mannosidase activity (Hosokawa et al. Citation2001). Therefore, EDEM1 was proposed to function as a quality control lectin that binds Man8B forms of N-glycan on misfolded proteins to promote ERAD (Hosokawa et al. Citation2001, 2003, Wang and Hebert Citation2003). Co-immunoprecipitation results have found that EDEM1 can differentiate between non-native and native proteins specifically binding to aberrant substrates (Hosokawa et al. Citation2006, Olivari et al. Citation2006, Cormier et al. Citation2009, Kosmaoglou et al. Citation2009). However, while EDEM1 efficiently bound to ERAD substrates lacking glycans, it did not accelerate the turnover of these non-glycosylated substrates (Cormier et al. Citation2009, Kosmaoglou et al. Citation2009). Furthermore, mutation of the mannosidase domain of EDEM1 still supports the accelerated degradation of ERAD substrates (Cormier et al. Citation2009, Termine et al. Citation2009, Hosokawa et al. Citation2010a). These results are suggestive of a bi-partite binding function for EDEM1 that involves both protein-protein interactions involving protein defects (perhaps exposed hydrophobic domains) and mannose trimmed glycans. While one form of binding might support interactions as probed by co-immunoprecipitation of lysed cells, apparently both binding forms must be engaged for EDEM1 to accelerate the turnover of an ERAD substrate.
There are a number of additional mysteries in regard to EDEM1 characterization and function. First, EDEM1 was original identified as a type II membrane protein that contained an N-terminal hydrophobic signal anchor sequence (Hosokawa et al. Citation2001). More recently a soluble form of EDEM1 has been observed generated by the cleavage of its N-terminal sequence (Olivari et al. Citation2005, Zuber et al. Citation2007). Second, EDEM1 has been shown to associate with calnexin, directly positioning it to extract ERAD cargo from the calnexin binding cycle (Oda et al. Citation2003). In contrast, morphological studies that explored the localization of endogenous EDEM1 found that it did not co-localize with calnexin and it could be found in the vesicle-like structures that exit the ER (Zuber et al. Citation2007). Third, the half-life of EDEM1 is short and autophagy or an autophagy-like pathway has been implicated in its rapid turnover (Cali et al. Citation2008, Le Fourn et al. Citation2009). The significance of this turnover route and accelerated rate is unknown but it may help explain why EDEM1 can be found in vesicles exiting the ER. Finally, the mannosidase-like domain of EDEM1 that has been proposed to mediate substrate recognition or trimming, has also been implicated in supporting EDEM1 binding to a downstream ER membrane adapter protein, SEL1L (Cormier et al. Citation2009). EDEM1 binding to endogenous SEL1L was diminished by mutating conserved residues in the mannosidase-like domain of EDEM1, or treatment with the mannosidase inhibitor kifunensine. These results suggest that the mannosidase-like domain may be deployed to target EDEM1-substrate complexes to ER membrane complexes involved in dislocation and ubiquitination. Future studies using purified proteins to recapitulate the substrate selection and targeting events would help to further understand these processes.
Mammalian EDEM1 has two paralogues termed EDEM2 and EDEM3 that are also upregulated by UPR and possess ER α-Mannosidase I homology domains (Mast et al. Citation2005, Olivari et al. Citation2005, Hirao et al. Citation2006). EDEM2 and EDEM3 have N-terminal cleavable signal sequences that generate soluble ER resident proteins. Only EDEM1 and EDEM3 have been found to be associated with increased mannose trimming when overexpressed in cells (Mast et al. Citation2005, Olivari et al. Citation2006, Hirao et al. Citation2006, Cormier et al. Citation2009, Hosokawa et al. Citation2010b). The overexpression of EDEM3 enhanced the mannose trimming of N-glycan of ERAD substrates to Man6 and Man7 (Hirao et al. Citation2006, Hosokawa et al. Citation2009). However, as the demonstration of mannosidase activity for isolated EDEM family members has not been demonstrated, it cannot be ruled out that EDEM interacting or stabilized proteins are responsible for the observed increase in cellular mannose trimming. In support of this possibility, ER α-Mannosidase I has been found to be stabilized by EDEM1 (Termine et al. Citation2009).
Quality control sorting receptors
Mannose trimming appears to generate an ERAD signal that must then be recognized by quality control receptors to target the defective cargo for ERAD. The family of possible mannose-dependent quality control receptors includes EDEM1 (as discussed above) and a group of ER resident MRH domain containing proteins that include Yos9p/OS-9 and XTP3-B. These MRH domain proteins bind to mannoses and interact with downstream ER membrane complexes involved in ERAD, implicating them in playing critical roles in the selection and delivery of defective secretory cargo to the ERAD pathway.
Yos9 was initially identified as an ERAD-related gene by genome screening of Saccharomyces cerevisiae as a Yos9 deletion strain stabilized CPY* (Buschhorn et al. Citation2004). It appeared to act specifically on glycosylated proteins since it did not stabilize non-glycosylated CPY* (Buschhorn et al. Citation2004, Kanehara et al. Citation2010). Its MRH domain is important for ERAD as mutating this domain suppressed CPY* turnover (Bhamidipati et al. Citation2005). Yos9p recognized ERAD substrates bearing Man8B and Man5 N-glycan (Szathmary et al. Citation2005). In contrast, frontal affinity chromatography analysis, a quantitative method developed to analyze carbohydrate-protein interactions, demonstrated that the MRH domain of Yos9p has high affinity for Man5 glycoforms but not Man8B (Quan et al. Citation2008). This later result suggested that Yos9p binds proteins that have been extensively trimmed by Mns1p and Htm1p to expose α1,6-linked mannose residues (Quan et al. Citation2008, Clerc et al. Citation2009). Since Yos9p also associated with non-glycosylated protein, it appears to possess both carbohydrate and protein-protein selection processes (Bhamidipati et al. Citation2005, Denic et al. Citation2006). Furthermore, Yos9p has also been found to associate with Hrd3p that nucleates the formation of an ERAD complex in the ER membrane (Carvalho et al. Citation2006, Denic et al. Citation2006, Gauss et al. Citation2006). Altogether, these results suggest that Yos9p is equipped with at least two functional domains: An MRH domain that supports aberrant glycoprotein recognition; and a protein-binding domain that binds to Hrd3p or misfolded proteins. These binding properties position Yos9p as an important link between substrate recognition and delivery to a downstream ER membrane ERAD complex.
There are two mammalian homologues of Yos9p called OS-9 and XTP3-B, which are both upregulated by UPR through Ire1/Xbp-1 activation (Bernasconi et al. Citation2008, Alcock and Swanton Citation2009). OS-9 and XTP3-B contain one and two MRH domains, respectively (Munro Citation2001). Like Yos9p, these proteins also appear to serve as quality control receptors that are able to selectively bind non-native substrates and sort them to the ERAD pathway through their association with an ER membrane adapter protein.
OS-9 has three spliced variants termed OS-9.1 through OS-9.3. Downregulation of OS-9 by RNAi stabilized glycosylated ERAD substrates; however, a similar fate was also observed upon overexpression (Bernasconi et al. Citation2008, Christianson et al. Citation2008, Hosokawa et al. Citation2009). This may be caused by the overexpressed protein binding to substrate but being ineffective in substrate presentation or downstream delivery due to the lack of a sufficient level of an associated factor. Overexpression of OS-9 may create an orphan subunit of the quality control receptor that produces a dominant negative effect on ERAD. The MRH domain of OS-9 has been shown to be critical for its role in ERAD (Hosokawa et al. Citation2009, Mikami et al. Citation2010). Recombinant human OS-9 MRH domain was recently found to associate preferentially with glycans missing C-arm mannose residues. Bound glycoforms include glycans that ranged from Glc1Man8 to the more extensively trimmed glycans such as Man4. These results were consistent with exposure of the α1,6-linked mannose on the C-arm being critical for OS-9 substrate recognition. Furthermore, OS-9 co-immunoprecipitated with mutant, but not wild type alpha-1-antitrypsin, demonstrating its ability to selectively bind defective substrates.
XTP3-B is a soluble ER protein with two MRH domains and two spliced variants (Hosokawa et al. Citation2008). The shorter variant is missing a small segment of the linker that connects the two MRH domains. The C-terminal MRH domain appears to be involved in substrate recognition as it is required for glycan-dependent binding to null Hong Kong alpha-1-antitrypsin (Yamaguchi et al. Citation2010). However, knockdown of XTP3-B does not affect the degradation of null Hong Kong alpha-1-antitrypsin, suggestive of redundant cellular factors being involved in its turnover (Christianson et al. Citation2008, Hosokawa et al. Citation2008). The C-terminal MRH domain of XTP3-B preferentially binds to Lec1 cells, which express proteins bearing Man5-6GlcNAc2 glycans on their surface (Yamaguchi et al. Citation2010). Inhibitory studies using various disaccharides revealed that Manα1,6Man efficiently inhibited substrate binding consistent with the second MRH domain of XTP3-B binding to terminally exposed α1,6-linked mannose residues. These results suggest that OS-9 and XTP3-B possess similar glycan binding requirements but as the two proteins are not associated with one another their roles appear to be distinct (Christianson et al. Citation2008). Interestingly, the simultaneous knockdown of both OS-9 and XTP3-B by RNAi specifically stabilized soluble proteins possessing luminal lesions (Bernasconi et al. Citation2010). Future studies will be required to determine if the two proteins are involved in redundant pathways or possess different binding requirements.
Similar to Yos9p, OS-9 and XTP3-B were found to associate with the mammalian Hrd3p homologue SEL1L, an ER membrane ERAD adapter protein (Christianson et al. Citation2008, Hosokawa et al. Citation2008, Mueller et al. Citation2008). Therefore in addition to selecting glycosylated ERAD substrates for degradation, these luminal quality control receptors also appear to support delivery of the defective substrates to the dislocation site in the ER membrane. The nature of the quality control receptor-adapter protein interaction is uncertain. It was reported that OS-9 and XTP3-B are recruited to the SEL1L complex by their MRH domains as mutation of the MRH domain of OS-9 and XTP3-B decreased their interaction with SEL1L but did not affect its association with ERAD substrates (Christianson et al. Citation2008) (). However, contradicting results are also available that suggest that OS-9 and SEL1L associate through protein-protein interactions () (Hosokawa et al. Citation2009, Yamaguchi et al. Citation2010). Similar uncertainty has also been observed with EDEM1 binding to SEL1L. The mannosidase-like domain of EDEM1 supports binding to the glycosylated SEL1L and not null Hong Kong alpha-1-antitrypsin as mutations of the mannosidase-like domain or kifunensine diminished EDEM1 binding to SEL1L and not to the ERAD substrates (Cormier et al. Citation2009).
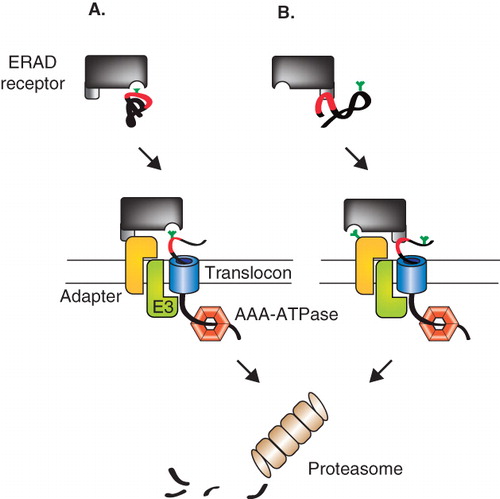
Figure 3. Two models for quality control receptor-substrate targeting to an ER membrane ERAD complex. (A) ERAD receptors recognize aberrant cargo displaying carbohydrate based ERAD signals using their sugar-binding domains (MRH or mannosidase-like domains). The receptor-substrate complex is then delivered to an ERAD complex in the ER membrane consisting of an adapter protein, an E3 ligase and a translocation channel or translocon. (B) In an alternative model for selection and delivery, the ERAD receptor selects client proteins based on the folding status of the protein through protein-protein interactions. Once a misfolded protein has been recognized the receptor uses its sugar-binding domain to dock to a glycan on the adapter protein to support delivery of the substrate to the ERAD complex.
Preparation of ERAD substrates for retrotranslocation
Once marked for ERAD and selected by quality control receptors for degradation, the non-native and possibly aggregated substrates need to be presented to an ERAD complex in a translocation competent form for their dislocation to the cytoplasm. As ERAD substrates can contain disulfide bonds, these events can require the protein-assisted reduction of disulfide bonds and unfolding.
The ER contains a family of oxidoreductases of which a number of these enzymes appear to play important roles in the reduction of intra- and inter-molecular disulfides to create ERAD substrates that are competent for dislocation. The extensively oxidized ERAD substrate BACE457 was found in a complex with PDI and BiP prior to dislocation to the cytoplasm (Molinari et al. Citation2002). Furthermore, PDI disassembled cholera toxin dimers to facilitate their retrotranslocation (Tsai et al. Citation2001). PDI has also been observed in a complex with Htm1p in yeast further implicating it in the ERAD process (Clerc et al. Citation2009, Sakoh-Nakatogawa et al. Citation2009). Interestingly, PDI stimulated the retrotranslocation of glycan and cysteine-free prepro (α) factor using mammalian and yeast microsome systems (Gillece et al. Citation1999, Wahlman et al. Citation2007). PDI appears to aid in substrate recognition and preparation for dislocation by both reduction, as well as by using its peptide binding properties to unfold misfolded and aggregated proteins.
The ER also contains a newly identified thioredoxin-domain containing flavoprotein called ERFAD (Riemer et al. Citation2009). The knockdown of ERFAD expression by RNAi stabilized ERAD substrate turnover and decreased the cellular level of poly-ubiquitinated proteins. ERFAD also interacted with a number of ERAD factors including OS-9, SEL1L and ERdj5 as shown by co-immunoprecipitation. ERdj5 possesses four thioredoxin domains, and a J-domain that supports the recruitment of BiP. ERdj5 was also identified as an EDEM1 interacting protein using a modified yeast two-hybrid and co-immunoprecipitation procedures (Ushioda et al. Citation2008). Its overexpression accelerated turnover of ERAD substrates by suppressing aggregation of misfolded proteins possessing non-native disulfide bonds. The activities of ERFAD, ERdj5, and their interacting partners, support them playing roles in the reduction and possibly unfolding of ERAD substrates so that they can be efficiently threaded through an ER translocon.
Adenine nucleotide regulated ER chaperones also appear to be involved in generating translocation competent ERAD substrates. BiP has been localized to ERAD complexes containing EDEM1 and ERdj5, as well as with XTP3-B (Christianson et al. Citation2008, Ushioda et al. Citation2008). In addition, the ER Hsp90 family member GRP94 was observed in a complex with OS-9 using mass spectrometry to analyze interacting partners of overexpressed tagged OS-9 (Christianson et al. Citation2008). The knockdown of GRP94 stabilized null Hong Kong alpha-1-antitrypsin. As both BiP and GRP94 are molecular chaperones, they may play a role in ERAD substrate recognition and/or their ATP-dependent binding cycle may aid in the unfolding of the misfolded substrates.
ER membrane ERAD adapter complexes
Once a protein has been deemed misfolded by an ERAD receptor, the misfolded protein needs to be targeted to a dislocation complex in a translocation competent form for the extraction of the misfolded protein to the cytoplasm. This ERAD complex appears to consist of a number of proteins that aid in the delivery, translocation and ubiquitination processes (Carvalho et al. Citation2006, Denic et al. Citation2006, Gauss et al. Citation2006, Christianson et al. Citation2008). Central to the formation of this ER membrane complex are adapter proteins that are integrated into the ER membrane and serve as the link between substrate recognition/delivery machinery and factors involved in dislocation and ubiquitination.
The yeast and mammalian ER contain ER membrane adapter proteins that possess a number of luminal-exposed tetratricopeptide repeat (TPR) domains (D'Andrea and Regan Citation2003). The yeast protein termed Hrd3p has nine TPR domains while the mammalian protein, SEL1L, contains a total of eleven. Structures of known TPR domains display helical folds comprised of two helices connected by a short linker. Adjacent TPR domains come together to form concave and convex surfaces, with protein-protein interactions generally occurring on the concave surface (Das et al. Citation1998). The well studied cytoplasmic TPR-containing protein Hop provides a link between Hsp70 and Hsp90 chaperones by using two separate clusters of TPR domains to recruit the individual chaperones to form a large protein folding complex (Scheufler et al. Citation2000). This cytoplasmic complex facilitates the passage of a maturing substrate from the Hsp70 to the Hsp90 chaperone system. Similarly, these ER membrane TPR domain proteins appear to create a connection between quality control receptors and dislocation/ubiquitination machinery to support the selective translocation of ERAD substrates to the cytoplasm for eventually destruction.
Affinity purification of different ERAD components followed by mass spectrometry was used to discover that Hrd3p resides in a large molecular weight complex, interacting with luminal (Yos9p), ER membrane (Usa1p, Hrd1p, Ubx2p) as well as cytosolic ERAD components (Cdc48p). This implicates Hrd3p serving as a scaffold or adapter protein that nucleates an ERAD translocation complex (Carvalho et al. Citation2006, Denic et al. Citation2006). Truncation studies with Hrd3p suggest that it binds Hrd1p and Yos9p through two distinct domains, implying that the TPR domains of Hrd3p direct distinct interactions with ERAD machinery (Gauss et al. Citation2006). Furthermore, Hrd3p also has the ability to directly bind misfolded proteins independent of Yos9p (Denic et al. Citation2006). The interaction between Hrd3p and Hrd1p may have multiple functions. First, Hrd3p appears to aid the delivery of misfolded proteins to the ER membrane E3 ligase, Hrd1p. Second, deletion of Hrd3p destabilized Hrd1p (Plemper et al. Citation1999, Gardner et al. Citation2000, Gauss et al. Citation2006), implying that Hrd3p might also be needed to stabilize the translocation complex, insuring that uncomplexed ligases do not accumulate.
The 11 TPR domains of SEL1L are arranged into three clusters comprised of four, five and two TPR domains moving from the N- to the C-terminus. SEL1L is N-linked glycosylated and has an N-terminal fibronectin type II domain (Biunno et al. Citation2006). SEL1L is known to interact with a large number of proteins including Hrd1, Herp, Derlin-1/2, EDEM1, OS-9 and XTP3-B (see below) (Lilley et al. Citation2006, Christianson et al. Citation2008, Hosokawa et al. Citation2008, Mueller et al. Citation2008, Cormier et al. Citation2009). Kopito and co-workers showed that SEL1L can bind to both OS-9 and XTP3-B without its transmembrane domain, but deletion of the two C terminal membrane proximal TPR domains abolished binding (Christianson et al. Citation2008). The role of SEL1L in ERAD was initially discovered by studying the cytomegalovirus-induced degradation of major histocompatibility complex class I heavy chains (Mueller et al. Citation2006). This study also demonstrated that knockdown of SEL1L stabilized a broader range of misfolded proteins.
As previously mentioned, both EDEM1 and XTP3-B have the ability to bind nonglycosylated misfolded proteins (Christianson et al. Citation2008, Hosokawa et al. Citation2008, Cormier et al. Citation2009, Kosmaoglou et al. Citation2009). Mutations in their respective MRH or mannosidase-like domains abolished binding between OS-9/XTP3-B/EDEM1 and SEL1L (Christianson et al. Citation2008, Cormier et al. Citation2009). This suggests that the MRH or mannosidase-like domains of the ERAD machinery help support SEL1L binding, rather than substrate recognition (). The glycans of SEL1L are all positioned either within or in close proximity to the TPR domain clusters, consistent with the possibility that the glycans and TPR domains work in concert to support protein interactions. Unlike for Hrd3p, SEL1L does not appear to directly bind ERAD clients (Christianson et al. Citation2008). Moreover the repertoire of SEL1L interacting proteins was recently expanded with the addition of UBC6e (an ER membrane anchor for an E2 ligase), UBXD8 (recruiter of the cytoplasmic AAA-ATPase p97) and AUP1 (ubiquitin-binding protein) (Mueller et al. Citation2008).
The Hrd3p complex in yeast also appears to contain an additional adapter protein termed Usa1p (Carvalho et al. Citation2006). Usa1p is needed for the degradation of luminal ERAD clients and serves as a linker for membrane proteins including Hrd1p and Der1p (Carvalho et al. Citation2006, Horn et al. Citation2009). The mammalian homolog of Usa1, Herp, is required for degradation of kappa light chain, a non-glycosylated subunit that is recognized by BiP rather than ER quality control lectin receptors (Okuda-Shimizu and Hendershot Citation2007). Like Usa1p, Herp is an ER membrane protein with both termini residing in the cytoplasm (Kokame et al. Citation2000). Interestingly, Herp was also shown to bind the H8 and S1 subunits of the proteasome, implying that Herp connects the translocation and ubiquitination processes to the proteasome (Okuda-Shimizu and Hendershot Citation2007). Together, these results indicated that adapter proteins play critical roles in the recruitment of ERAD machinery within the lumen, cytoplasm, as well as the ER membrane.
Translocation and ubiquitination
The final ER events for ERAD include the translocation of the ERAD substrate across the ER membrane and its poly-ubiquitination at the cytoplasmic face of the ER membrane by membrane localized E3 ligases. Machinery involved in these important processes appears to be components of the large membrane protein complex nucleated by the ER membrane adapter proteins (). The organization of these complexes insures the efficient transfer of ERAD substrates from quality control receptors, to translocons and their ubiquitination upon arrival in the cytoplasm.
Figure 4. ER membrane ERAD complexes. Proteins that reside in the yeast and mammalian ERAD translocation complex are designated.
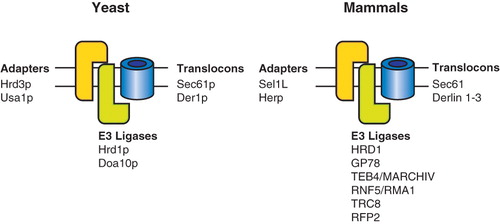
The polytopic ER membrane protein Derlin-1 and its yeast homologue Der1p associate with the SEL1L/HRD1 and Hrd3p/Hrd1p adapter protein complexes, respectively (Lilley and Ploegh Citation2005, Schulze et al. Citation2005, Ye et al. Citation2005, Carvalho et al. Citation2006). The human cytomegalovirus initiated turnover of major histocompatibility class I heavy chain involves the use of Derlin proteins, as Derlin-1 was discovered as an US11-interacting protein that accelerated the degradation of heavy chain (Lilley and Ploegh Citation2004). Two additional homologues of Derlin have been discovered termed Derlin-2 and Derlin-3. The overexpression of the Derlin family members accelerated the degradation of ERAD substrates, whereas its knockdown stabilized them (Oda et al. Citation2006, Sun et al. Citation2006). Additional studies will be required to determine if Derlins serve as translocation pores, as this role is largely speculative and based on their topology that includes four putative transmembrane domains.
The Sec61 translocon that is responsible for the insertion of proteins into the ER has also been proposed to play a role in the translocation of ERAD substrates in the reverse direction (Wiertz et al. Citation1996). This demonstration is complicated by the need to delineate the protein import and export processes. However, yeast strains possessing mutations in Sec61p have been described that support efficient translocation into the ER but are deficient in the retrotranslocation of proteins out of the ER (Pilon et al. Citation1997, Plemper et al. Citation1997, Citation1998, Scott and Schekman Citation2008). Furthermore, yeast and mammalian ERAD substrates have been observed to associate with Sec61 during late stages of quality control (Wiertz et al. Citation1996, Pilon et al. Citation1997, Plemper et al. Citation1997). Future studies will be required to determine the range of proteins that can serve as retrotranslocons, and if each of the translocons supports the transit of different types of substrates. Additionally, while the AAA-ATPase p97 (Cdc48p in yeast) has been implicated in driving the translocation of the substrate once the termini of the substrate reaches the cytoplasm, it is unclear what the mechanism is for the initial threading of the ERAD substrate in through the pore. Cotranslational translocation is aided by chain elongation driven by the ribosome, which is nestled on the translocon. However, a post-translocation event would appear to require an initial push from the lumen for the chain to reach the cytoplasmic AAA-ATPase involved in pulling the chain into the cytoplasm.
During the arrival in the cytoplasm, ERAD substrates are rapidly ubiquitinated by a family of ER localized E3 enzymes. The topology of the ERAD substrate and the location of the defect appear to determine the identity of its modifier. In yeast, membrane proteins possessing cytoplasmic defects or soluble cytoplasmic proteins were modified by Doa10p (Vashist and Ng Citation2004). In contrast, soluble or membrane proteins possessing luminal lesions used Hrd1p (Carvalho et al. Citation2006, Denic et al. Citation2006). The transmembrane region of Hrd1p also appears to support the direct recognition of proteins with defects that lie within the membrane (Sato et al. Citation2009). Altogether, Hrd1p appears to manage the formation of a number of ERAD complexes that support the processing of different substrate classes (Kanehara et al. Citation2010).
In mammalian cells, the family of E3 ligases involved in ERAD has expanded beyond the small set of yeast modifiers. In addition to the yeast Hrd1p homologue HRD1, mammalian E3 ligases currently include GP78, RNF5/RMA1, TEB4/MARCHIV, TRC8 and RFP2 (Fang et al. Citation2001, Hassink et al. Citation2005, Lerner et al. Citation2007, Morito et al. Citation2008, Stagg et al. Citation2009). The selectivity of many of the ER E3 ligases is poorly defined. The selection process appears to differ from the yeast system where Hrd1p ubiquitinates substrates with defects within the membrane or luminal compartments (Vashist and Ng Citation2004). Recent results suggest that soluble ERAD substrates that are dependent on HRD1, can be rendered HRD1-independent by simply tethering them to the membrane with a single hydrophobic domain (Bernasconi et al. Citation2010). The change in topology apparently alters the E3 ligase involved in directing turnover. The larger spectrum of ERAD substrates found in multicellular systems has resulted in the expansion of E3 ligases and diversification of the selection process. In some cases, the association of ligases with specific adapter complexes appears to dictate the substrate selection process, whereas for other ligases substrates may be selected directly by the ligase.
Conclusion
The quality control process evaluates mutant proteins that are defective and targets them for degradation. In addition, as one third of the genome products traverse the secretory pathway and maturation is an error prone process, the quality control machinery must also test all the secretory client proteins. While many quality control components have been identified along with the complexes in which they reside, how this relatively small group of proteins efficiently screens the thousands of different cargo proteins is still poorly understood. In the cytoplasm, hundreds of E3 ligases are responsible for the recognition, modification and turnover of cytosolic factors (Deshaies and Joazeiro Citation2009). Since recognition and modification of secretory pathway proteins occurs in two separate locations, this would appear to provide an additional layer of complexity; however, the machinery involved appears to be simplified. Future studies will be required to determine how a glycan-based quality control process can efficiently manage such a large group of diverse customers that includes wild type and mutant proteins, misfolded or unassembled cargo, and factors that are degraded through regulated proteolysis.
Declaration of interest: This work was supported by US Public Health grants CA79864 and GM086874 (DNH) and The Uehara Memorial Foundation (TT). In addition, JCS was partially supported by an NIH Chemistry-Biology Interface training grant (T32GM00815). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alcock F, Swanton E. 2009. Mammalian OS-9 is upregulated in response to endoplasmic reticulum stress and facilitates ubiquitination of misfolded glycoproteins. J Mol Biol 385:1032–1042.
- Apweiler R, Hermjakob H, Sharon N. 1999. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473:4–8.
- Avezov E, Frenkel Z, Ehrlich M, Herscovics A, Lederkremer GZ. 2008. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-Glycan trimming to Man5 6GlcNAc2 in glycoprotein ER-associated degradation. Mol Biol Cell 19:216–225.
- Ayalon-Soffer M, Shenkman M, Lederkremer GZ. 1999. Differential roles of mannose and glucose trimming in the ER degradation of asialoglycoprotein receptor subunits. J Cell Sci 112:3309–3318.
- Barile M, Pisitkun T, Yu MJ, Chou CL, Verbalis MJ, Shen RF, Knepper MA. 2005. Large scale protein identification in intracellular aquaporin-2 vesicles from renal inner medullary collecting duct. Mol Cell Proteomics 4:1095–1106.
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. 2010. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol 188:223–235.
- Bernasconi R, Pertel T, Luban J, Molinari M. 2008. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: Inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem 283:16446–16454.
- Bhamidipati A, Denic V, Quan EM, Weissman JS. 2005. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell 19:741–751.
- Biunno I, Cattaneo M, Orlandi R, Canton C, Biagiotti L, Ferrero S, Barberis M, Pupa SM, Scarpa A, Menard S. 2006. SEL1L a multifaceted protein playing a role in tumor progression. J Cell Physiol 208:23–38.
- Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething M-JH. 1993. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75:717–728.
- Buschhorn BA, Kostova Z, Medicherla B, Wolf DH. 2004. A genome-wide screen identifies Yos9p as essential for ER-associated degradation of glycoproteins. FEBS Lett 577:422–426.
- Cabral CM, Liu Y, Sifers RN. 2001. Dissecting glycoprotein quality control in the secretory pathway. Tren Biochem Sci 26:619–624.
- Cali T, Galli C, Olivari S, Molinari M. 2008. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 371:405–410.
- Caramelo JJ, Castro OA, Alonso LG, de Prat-Gay G, Parodi AJ. 2003. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Acad Sci USA 100:86–91.
- Caramelo JJ, Castro OA, de Prat-Gay G, Parodi AJ. 2004. The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediates. J Biol Chem 279:46280–46285.
- Carvalho P, Goder V, Rapoport TA. 2006. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126:361–373.
- Chen W, Helenius J, Braakman I, Helenius A. 1995. Cotranslational folding and calnexin binding of influenza hemagglutinin in the endoplasmic reticulum. Proc Natl Acad Sci USA 92:6229–6233.
- Chevet E, Wong HN, Gerber D, Cochet C, Fazel A, Cameron PH, Gushue JN, Thomas DY, Bergeron JJ. 1999. Phosphorylation by CK2 and MAPK enhances calnexin association with ribosomes. EMBO J 18:3655–3666.
- Christianson JC, Shaler TA, Tyler RE, Kopito RR. 2008. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 10:272–282.
- Chung DH, Ohashi K, Watanabe M, Miyasaka N, Hirosawa S. 2000. Mannose trimming targets mutant alpha 2 plasmin inhibitor for degradation by the proteasome. J Biol Chem 275:4981–4987.
- Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, Aebi M. 2009. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol 184:159–172.
- Coe H, Jung J, Groenendyk J, Prins D, Michalak M. 2010. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem 285:6725–6738.
- Cormier JH, Tamura T, Sunryd JC, Hebert DN. 2009. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Molec Cell 34:627–633.
- D'Andrea LD, Regan L. 2003. TPR proteins: The versatile helix. Trends Biochem Sci 28:655–662.
- Daniels R, Kurowski B, Johnson AE, Hebert DN. 2003. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell 11:79–90.
- Das AK, Cohen PW, Barford D. 1998. The structure of the tetratricopeptide repeats of protein phosphatase 5: Implications for TPR-mediated protein-protein interactions. EMBO J 17:1192–1199.
- Denic V, Quan EM, Weissman JS. 2006. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126:349–359.
- Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, Stamp G, Owen MJ. 2002. Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 22:7398–7404.
- Deprez P, Gautschi M, Helenius A. 2005. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell 19:183–195.
- Deshaies RJ, Joazeiro CA. 2009. RING domain E3 ubiquitin ligases. Annu Rev Biochem 78:399–434.
- Ermonval M, Kitzmuller C, Mir AM, Cacan R, Ivessa NE. 2001. N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology 11:565–576.
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. 2001. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA 98:14422–14427.
- Frenkel Z, Gregory W, Kornfeld S, Lederkremer GZ. 2003. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6-5GlcNAc2. J Biol Chem 278:34119–33424.
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. 2000. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol 151:69–82.
- Gauss R, Jarosch E, Sommer T, Hirsch C. 2006. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol 8:849–854.
- Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, Lesimple S, Nagaya H, Roy L, Gosline SJ, Hallett M, Paiement J, Kearney RE, Nilsson T, Bergeron JJ. 2006. Quantitative proteomics analysis of the secretory pathway. Cell 127:1265–1281.
- Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Romisch K. 1999. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J Cell Biol 147:1443–1456.
- Gnann A, Riordan JR, Wolf DH. 2004. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell 15:4125–4135.
- Gong Q, Keeney DR, Molinari M, Zhou Z. 2005. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem 280:19419–19425.
- Hassink G, Kikkert M, van Voorden S, Lee SJ, Spaapen R, van Laar T, Coleman CS, Bartee E, Fruh K, Chau V, Wiertz E. 2005. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J 388:647–655.
- Hebert DN, Foellmer B, Helenius A. 1995. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81:425–433.
- Hebert DN, Foellmer B, Helenius A. 1996. Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J 15:2961–2968.
- Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. 1997. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol 139:613–623.
- Helenius A. 1994. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell 5:253–265.
- Herscovics A, Romero PA, Tremblay LO. 2002. The specificity of the yeast and human class I ER alpha1,2-mannosidase involved in ER quality control is not as strict as previously reported. Glycobiology 12:14G–15G.
- Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, Nagata K, Hosokawa N. 2006. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J Biol Chem 281:9650–9658.
- Hirayama H, Seino J, Kitajima T, Jigami Y, Suzuki T. 2010. Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in Saccharomyces cerevisiae. J Biol Chem 285:12390–12404.
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. 2009. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell 36:782–793.
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. 2009. Human OS-9, a lectin required for glycoprotein ERAD, recognizes mannose-trimmed N-glycans. J Biol Chem 20:310–321.
- Hosokawa N, Kamiya Y, Kato K. 2010a. The role of MRH domain-containing lectins in ERAD. Glycobiology 284:17061–17068.
- Hosokawa N, Tremblay LO, Sleno B, Kamiya Y, Wada I, Nagata K, Kato K, Herscovics A. 2010b. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 20:567–575.
- Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. 2003. Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J Biol Chem 278:26287–26294.
- Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. 2001. A novel ER a-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep 2:415–422.
- Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. 2008. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem 283:20914–20924.
- Hosokawa N, Wada I, Natsuka Y, Nagata K. 2006. EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-antitrypsin. Genes Cells 11:465–476.
- Jakob CA, Burda P, Roth J, Aebi M. 1998. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J Cell Biol 142:1223–1233.
- Kanehara K, Xie W, Ng DT. 2010. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J Cell Biol 188:707–716.
- Kearse KP, Roberts JL, Munitz TI, Wiest DL, Nakayama T, Singer A. 1994. Developmental regulation of ab T cell antigen receptor expression results from differen stability of nascent TCRa proteins within the endoplasmic reticulum and mature T cells. EMBO J 13:4504–4514.
- Keith N, Parodi AJ, Caramelo JJ. 2005. Glycoprotein tertiary and quaternary structures are monitored by the same quality control mechanism. J Biol Chem 280:18138–18141.
- Keller SH, Lindstrom J, Taylor P. 1998. Inhibition of glucose trimming with castanospermine reduces calnexin association and promotes proteasomal degradation of the a-subunit of the nicotinic acetylcholine receptor. J Biol Chem 273:17064–17072.
- Kitzmuller C, Caprini A, Moore SE, Frenoy JP, Schwaiger E, Kellermann O, Ivessa NE, Ermonval M. 2003. Processing of N-linked glycans during endoplasmic-reticulum-associated degradation of a short-lived variant of ribophorin I. Biochem J 376:687–696.
- Kokame K, Agarwala KL, Kato H, Miyata T. 2000. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem 275:32846–32853.
- Kosmaoglou M, Kanuga N, Aguila M, Garriga P, Cheetham ME. 2009. A dual role for EDEM1 in the processing of rod opsin. J Cell Sci 122:4465–4472.
- Le Fourn V, Gaplovska-Kysela K, Guhl B, Santimaria R, Zuber C, Roth J. 2009. Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell Mol Life Sci 66:1434–1445.
- Lederkremer GZ. 2009. Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol 19:515–523.
- Lerner M, Corcoran M, Cepeda D, Nielsen ML, Zubarev R, Ponten F, Uhlen M, Hober S, Grander D, Sangfelt O. 2007. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol Biol Cell 18:1670–1682.
- Lilley BN, Gilbert JM, Ploegh HL, Benjamin TL. 2006. Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J Virol 80:8739–8744.
- Lilley BN, Ploegh HL. 2004. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429:834–840.
- Lilley BN, Ploegh HL. 2005. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 102:14296–14301.
- Liu Y, Choudhury P, Cabral CM, Sifers RN. 1999. Oligosaccharide modification in the early secretory pathway directs the selection of a misfolded glycoprotein for degradation by the proteasome. J Biol Chem 274:5861–5867.
- Mackeen MM, Almond A, Deschamps M, Cumpstey I, Fairbanks AJ, Tsang C, Rudd PM, Butters TD, Dwek RA, Wormald MR. 2009. The conformational properties of the Glc3Man unit suggest conformational biasing within the chaperone-assisted glycoprotein folding pathway. J Mol Biol 387:335–347.
- Mast SW, Diekman K, Karaveg K, Davis A, Sifers RN, Moremen KW. 2005. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15:421–436.
- Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause K-H, Opas M, MacLennan DH, Michalak M. 1999. Calreticulin is essential for cardiac development. J Cell Biol 144:857–868.
- Mikami K, Yamaguchi D, Tateno H, Hu D, Qin SY, Kawasaki N, Yamada M, Matsumoto N, Hirabayashi J, Ito Y, Yamamoto K. 2010. The sugar-binding ability of human OS-9 and its involvement in ER-associated degradation. Glycobiology 20:310–321.
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. 2003. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299:1397–1400.
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. 2002. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol 158:247–257.
- Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. 2005. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell 20:503–512.
- Molinari M, Helenius A. 2000. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 288:331–333.
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K. 2008. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell 19:1328–1336.
- Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. 2008. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA 105:12325–12330.
- Mueller B, Lilley BN, Ploegh HL. 2006. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol 175:261–270.
- Munro S. 2001. The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr Biol 11:R499–501.
- Oda Y, Hosokawa N, Wada I, Nagata K. 2003. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299:1394–1397.
- Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. 2006. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol 172:383–393.
- Okuda-Shimizu Y, Hendershot LM. 2007. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell 28:544–554.
- Olivari S, Cali T, Salo KE, Paganetti P, Ruddock LW, Molinari M. 2006. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem Biophys Res Commun 349:1278–1284.
- Olivari S, Galli C, Alanen H, Ruddock L, Molinari M. 2005. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J Biol Chem 280:2424–2428.
- Oliver JD, van der Wal FJ, Bulleid NJ, High S. 1997. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 275:86–88.
- Ou WJ, Cameron PH, Thomas DY, Bergeron JJ. 1993. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature 364:771–776.
- Pearse BR, Gabriel L, Wang N, Hebert DN. 2008. A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. J Cell Biol 181:309–320.
- Pearse BR, Hebert DN. 2010. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim Biophys Acta 1803:684–693.
- Pearse BR, Tamura T, Sunryd JC, Grabowski GA, Kaufman RJ, Hebert DN. 2010. The role of UDP-Glc: glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate. J Cell Biol 189:829–841.
- Peterson JR, Ora A, Nguyen Van P, Helenius A. 1995. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol Biol Cell 6:1173–1184.
- Pilon M, Schekman R, Romisch K. 1997. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J 16:4540–4548.
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. 1997. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388:891–895.
- Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH. 1999. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci 112:4123–4134.
- Plemper RK, Egner R, Kuchler K, Wolf DH. 1998. Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J Biol Chem 273:32848–32856.
- Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. 2008. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell 32:870–877.
- Rajagopalan S, Xu Y, Brenner MB. 1994. Retention of unassembled components of integral membrane proteins by calnexin. Science 263:387–390.
- Riemer J, Appenzeller-Herzog C, Johansson L, Bodenmiller B, Hartmann-Petersen R, Ellgaard L. 2009. A luminal flavoprotein in endoplasmic reticulum-associated degradation. Proc Natl Acad Sci USA 106:14831–14836.
- Sakoh-Nakatogawa M, Nishikawa S, Endo T. 2009. Roles of protein-disulfide isomerase-mediated disulfide bond formation of yeast Mnl1p in endoplasmic reticulum-associated degradation. J Biol Chem 284:11815–11825.
- Sato BK, Schulz D, Do PH, Hampton RY. 2009. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell 34:212–222.
- Schallus T, Jaeckh C, Feher K, Palma AS, Liu Y, Simpson JC, Mackeen M, Stier G, Gibson TJ, Feizi T, Pieler T, Muhle-Goll C. 2008. Malectin: A novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol Biol Cell 19:3404–3414.
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. 2000. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210.
- Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. 2005. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol 354:1021–1027.
- Scott DC, Schekman R. 2008. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol 181:1095–1105.
- Solda T, Garbi N, Hammerling GJ, Molinari M. 2006. Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J Biol Chem 281:6219–6226.
- Sousa M, Parodi AJ. 1995. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc: glycoprotein glucosyltransferase. EMBO J 14:4196–4203.
- Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM, Lehner PJ. 2009. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol 186:685–692.
- Stigliano ID, Caramelo JJ, Labriola CA, Parodi AJ, D'Alessio C. 2009. Glucosidase II beta subunit modulates N-glycan trimming in fission yeasts and mammals. Mol Biol Cell 20:3974–3984.
- Su K, Stoller T, Rocco J, Zemsky J, Green R. 1993. Pre-Golgi degradation of yeast prepro-a-factor in a mammalian cell. J Biol Chem 268:14301–14309.
- Sun F, Zhang R, Gong X, Geng X, Drain PF, Frizzell RA. 2006. Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J Biol Chem 281:36856–36863.
- Svedine S, Wang T, Halaban R, Hebert DN. 2004. Carbohydrates act as sorting determinants in ER-associated degradation of tyrosinase. J Cell Sci 117:2937–2949.
- Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. 2005. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell 19:765–775.
- Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY. 2003. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep 4:405–411.
- Termine DJ, Moremen KW, Sifers RN. 2009. The mammalian UPR boosts glycoprotein ERAD by suppressing the proteolytic downregulation of ER mannosidase I. J Cell Sci 122:976–984.
- Totani K, Ihara Y, Matsuo I, Tsujimoto T, Ito Y. 2009. The recognition motif of the glycoprotein-folding sensor enzyme, UDP-Glc: Glycoprotein Glucosyltransferase. Biochemistry 48:2933–2940.
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. 2000. Functional and genomic analyses reveal an essential coordination between unfolded protein response and ER-associated degradation. Cell 101:249–258.
- Trombetta ES, Helenius A. 2000. Conformational requirements for glycoprotein reglucosylation in the endoplasmic reticulum. J Cell Biol 148:1123–1129.
- Tsai B, Rodighiero C, Lencer WI, Rapoport TA. 2001. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104:937–948.
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. 2008. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321:569–572.
- Vashist S, Ng DT. 2004. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165:41–52.
- Vassilakos A, Cohen-Doyle MF, Peterson PA, Jackson MR, Williams DB. 1996. The molecular chaperone calnexin facilitates folding and assembly of class I histocompatibility molecules. EMBO J 15:1495–1506.
- Wada I, Kai M, Imai S, Sakane F, Kanoh H. 1997. Promotion of transferrin folding by cyclic interactions with calnexin and calreticulin. EMBO J 16:5420–5432.
- Wahlman J, Demartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. 2007. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 129:943–955.
- Wang N, Daniels R, Hebert DN. 2005. The cotranslational maturation of the type I membrane glycoprotein tyrosinase: The heat shock protein 70 system hands off to the lectin-based chaperone system. Mol Biol Cell 16:3740–3752.
- Wang N, Glidden EJ, Murphy SR, Pearse BR, Hebert DN. 2008. The cotranslational maturation program for the type II membrane glycoprotein influenza neuraminidase. J Biol Chem 283:33826–33837.
- Wang T, Hebert DN. 2003. EDEM an ER quality control receptor. Nature Struct Biol 10:319–321.
- Watanabe T, Totani K, Matsuo I, Maruyama J, Kitamoto K, Ito Y. 2009. Genetic analysis of glucosidase II beta-subunit in trimming of high-mannose-type glycans. Glycobiology 19:834–840.
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. 1996. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction [see comments]. Nature 384:432–438.
- Wu Y, Termine DJ, Swulius MT, Moremen KW, Sifers RN. 2007. Human endoplasmic reticulum mannosidase I is subject to regulated proteolysis. J Biol Chem 282:4841–4849.
- Yamaguchi D, Hu D, Matsumoto N, Yamamoto K. 2010. Human XTP3-B binds to alpha1-antitrypsin variant null (Hong Kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology 20:348–355.
- Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. 2005. Inaugural article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 102:14132–14138.
- Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJ, Thomas DY. 1998. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem 273:6009–6012.
- Zhang J-X, Braakman I, Matlack KES, Helenius A. 1997. Quality control in the secretory pathway: The role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol Biol Cell 8:1943–1954.
- Zuber C, Cormier JH, Guhl B, Santimaria R, Hebert DN, Roth J. 2007. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc Natl Acad Sci USA 104:4407–4412.
- Zuber C, Fan JY, Guhl B, Parodi A, Fessler JH, Parker C, Roth J. 2001. Immunolocalization of UDP-glucose:glycoprotein glucosyltransferase indicates involvement of pre-Golgi intermediates in protein quality control. Proc Natl Acad Sci USA 98:10710–10715.