Abstract
Here we review a novel class of delivery vehicles based on pH-sensitive, moderately polar membrane peptides, which we call pH (Low) Insertion Peptides (pHLIPs), that target cells located in the acidic environment found in many diseased tissues, including tumours. Acidity targeting by pHLIPs is achieved as a result of helix formation and transmembrane insertion. In contrast to the earlier technologies based on cell-penetrating peptides, pHLIPs act as monomeric membrane-inserting peptides that translocate one terminus across a membrane into the cytoplasm, while the other terminus remains in the extracellular space, locating the peptide in the membrane lipid bilayer. Therefore pHLIP has a dual delivery capability: it can tether cargo molecules or nanoparticles to the surfaces of cells in diseased tissues and/or it can move a cell-impermeable cargo molecule across the membrane into the cytoplasm. The source of energy for moving polar molecules attached to pHLIP through the hydrophobic layer of a membrane bilayer is the membrane-associated folding of the polypeptide. A drop in pH leads to the protonation of negatively charged residues (Asp or Glu), which enhances peptide hydrophobicity, increasing the affinity of the peptide for the lipid bilayer and triggering peptide folding and subsequent membrane insertion. The process is accompanied by the release of energy that can be utilized to move cell-impermeable cargo across a membrane. That the mechanism is now understood, and that targeting of tumours in mice has been shown, suggest a number of future applications of the pHLIP technology in the diagnosis and treatment of disease.
Introduction
Medical science constantly seeks to improve the diagnosis and treatment of disease, and new delivery technologies may contribute in several ways, including:
Targeted therapy: Selective delivery of therapeutic and imaging agents to diseased tissue, thereby increasing the effective concentration of these agents and reducing their accumulation in healthy tissue.
Improved routes of drug administration: Encapsulation of therapeutic agents to improve pharmacokinetic properties of a drug.
Locally activated therapy: Activation of a targeted therapeutic agent by external effectors.
Multi-functionality: Simultaneous targeted delivery of a therapeutic agent and an imaging probe to monitor drug distribution.
New approaches to drug design: Use of a new class of therapeutic agents: cell-impermeable molecules that are translocated into cells only in diseased tissue while not affecting healthy cells.
A therapy employing any or all of these concepts might exhibit much higher efficacy and/or significantly reduced side effects. Such improvements are especially needed for cancer treatment, since the majority of anti-cancer drugs are poisons that damage normal cells. In the following, we place special emphasis on cancer, but it should be recognized that other diseased tissues might be treated using the strategies we discuss.
Nanosized carriers for improved route of drug administration and multi-functionality
Various nanosized drug-delivery vehicles, such as organic, metallic or semiconductor nanoparticles, liposomes, micelles, viral particles, polymers and dendrimers are designed to address the limitations of conventional drug delivery systems, including low aqueous solubility, poor bioavailability and low therapeutic indices stemming from insufficient drug concentration at disease sites (Davis et al. Citation2008, Gindy and Prud'homme Citation2009). The principle of drug encapsulation is extremely important, since it may create an opportunity to use many amphiphilic or polar drugs that have established activities but cannot be used since they do not passively cross a cell membrane. Another advantage is multi-functionality: Both therapeutic and imaging agents can be loaded at the same time in one nanoparticle to monitor the amount and location of a therapeutic administration, using clinical imaging methods. Among nanosized drug carriers are self assembling lipid-containing systems such as liposomes and micelles, which deliver molecules trapped inside or within their hydrophobic phospholipid bilayer (Semalty et al. Citation2009). Viruses have also been envisaged as nanoparticle vectors suitable for drug delivery, vaccines, and gene therapy due to their regular geometries, well characterized surface properties, and nanoscale dimensions (Lin and Nemunaitis Citation2004, Aghi and Martuza Citation2005, Everts and van der Poel Citation2005). At the same time, despite extensive investigation of dozens of viral vectors for cancer treatment, and promising results in clinical trials, the FDA has not approved any virus-based therapeutics, because of toxicities widely reported in gene therapy. Various polymer molecules are traditionally employed as drug delivery systems (Moghimi Citation2006). Formulations using dendrimers, hyper-branched synthetic macromolecules with controllable sizes and shapes, for drug delivery may have advantages over traditional polymeric systems (Samad et al. Citation2009, Tekade et al. Citation2009).
Local activation therapy
An evolving strategy is to target an inactive entity to a tissue, and then activate it with a signal, reducing incidental damage to a patient below the level attainable by either a targeting strategy or an activating signal alone. Such synergistic binary approaches might use activation by electromagnetic radiation, ultrasound, neutrons or other effectors (Kankaanranta et al. Citation2007, Pisarev et al. Citation2007, O'Connor et al. Citation2009, Schroeder et al. Citation2009). These are promising directions for the development of therapeutic approaches with restricted side effects. Nanotechnology has led to the fabrication of inorganic nanomaterials, including nanocrystals, nanotubes and nanowires, with physical properties that appear promising for biological and medical applications. For example, carbon nanotubes, nanoshells and gold nanorods have attracted attention due to their strong absorbance in the near infrared (NIR) window and release of energy in the form of heat, which can induce irreversible local damage to the cells in a tumour (O'Connell et al. Citation2002, Hainfeld et al. Citation2004, Morris et al. Citation2006). Hyperthermia can also be stimulated by using magnetic field fluctuations to heat magnetic-nanoparticles (Ito et al. Citation2005, Duguet et al. Citation2006, Latorre and Rinaldi Citation2009). Neutron capture therapy based on the irradiation of boron-10 or gadolinium clusters with thermal neutrons, generating short range high linear energy ionizing particles, provides a means to deliver radiation locally at the cellular level (Kankaanranta et al. Citation2007, Pisarev et al. Citation2007, Mitin et al. Citation2009). Another approach is to use NIR light to activate light sensitive molecules to produce free radicals, which can destroy cancer cells (photodynamic therapy) (Palumbo Citation2007, O'Connor et al. Citation2009). Interaction of gold nanoparticles with keV radiation of an appropriate energy can generate Auger and photo-electrons inducing death of the cells in the vicinity of a gold nanoparticle (Hainfeld et al. Citation2004). Light or ultrasound can also be used to enhance drug release at a disease site (Liapi and Geschwind, Citation2007, Schroeder et al. Citation2009). Thus, there are many strategies that might be used if selective delivery to a diseased tissue can be accomplished. However, finding the means to accomplish the selective delivery to tumours or other tissues still remains a challenging problem, as discussed below.
Disease-specific delivery coupled with local activation would allow: (i) Accumulating and, therefore, increasing the effective concentration of therapeutic or diagnostic agents in a diseased area, and so (ii) reducing the side-effects associated with treatment by reducing the targeting of normal cells. Adding the dimension of local activation would further improve the protection of normal tissue.
Cancer biomarkers and traditional targeting strategies
A great deal of effort has been devoted to the design and development of tumour selective agents to improve visualization and treatment of cancerous tissue. Traditionally, receptors and enzymes overexpressed in cancer cells are considered to be cancer biomarkers, serving as indicators of the change in physiologic state during the disease process (Srinivas et al. Citation2001, Hanke et al. Citation2004, Janssens et al. Citation2004), and so there has been a focus on peptides and antibody fragments directed toward cell surface receptors (Goldsmith Citation1997, Freimark et al. Citation2007). Monoclonal antibodies were initially thought to be the most promising candidates for specific targeting strategies; however, because of problems associated with their specificity and high molecular weight, clinically successful developments have proven difficult (Blättler and Chari Citation2001). Only over the last few years has advanced antibody engineering technology enabled therapeutic concepts based on antibodies and conjugates thereof to successfully enter clinical practice (Carter Citation2001, Payne Citation2003). Antibodies and their fragments have been used to map the expression or overexpression of tumour-related proteins, such as prostate-specific membrane antigen (Polascik et al. Citation1999), human epidermal growth factor receptor-2 (HER2) (Moasser Citation2007); carcinoembryonic antigen (Muxi et al. Citation1999, Lu et al. Citation2007), TAG-72 (Muxi et al. Citation1999), Ep-CAM (de Bono et al. Citation2004) and others. However, a number of complications still vex antibody applications, such as purity, immunogenicity, slow diffusion in tissues, plasma clearance, and production difficulties (Blättler and Chari Citation2001).
An attractive, related direction is the development of low molecular weight peptides for rapid tumour targeting. In contrast to antibodies, peptides can be easily synthesized, modified and stabilized to obtain optimized pharmacokinetic parameters (Lister-James et al. Citation1997, Signore et al. Citation2001). Usually they are not immunogenic and can have high receptor affinity. The most developed and widely accepted are somatostatin analogs introduced to visualize various somatostatin receptor-positive tumours (Krenning et al. Citation1999, Buchsbaum Citation2004). A number of other peptides targeting various receptors expressed in cancer cells have recently been tested for tumour diagnosis as well (Signore et al. Citation2001, Ma et al. Citation2007).
Tumour acidosis
The most important limitation of specific cancer cell receptor targeting is the heterogeneity of human cancers (Jeffrey et al. Citation2005). Recent studies of gene expression in cancer cells indicate that a number of genes are up- and down-regulated, and that cells in a tumour are heterogeneous. It is therefore problematic to rely on any single tumour biomarker even for one type of cancer (Bild et al. Citation2006). Using tumour acidity may be an alternative, since it is well established that salient features of the microenvironment of solid tumours include hypoxia and extracellular acidity (Griffiths Citation1991, Gatenby and Gillies Citation2008). These factors contribute to the selection of the cancerous phenotype, and also to the progression from benign to malignant tumours. Acidosis is associated with tumour development both at very early and at advanced stages (Gillies et al. Citation1994, Raghunand et al. Citation1999b). Rapidly proliferating cancer cells become partially anaerobic, leading to the elevation of glycolysis in response to hypoxia (Pasteur effect) (Krebs Citation1972). Hypoxia and acidity are partly a result of the chaotic and heterogeneous microvasculature structure of solid tumours, where the oxygen concentration decreases with distance from a capillary (Gillies et al. Citation1999, Schornack and Gillies Citation2003). Hypoxia and low blood supply are involved in cancer progression, but they are not the only mechanism responsible for the development of an acidic environment within solid tumours. A hallmark of malignant cancers is an elevated glucose uptake even under normal oxygen conditions, known as ‘aerobic glycolysis’ or the Warburg effect (Warburg et al. Citation1927). Cells exhibiting a Warburg effect catabolize glucose at a high rate (Newell et al. Citation1993, Gillies et al. Citation2008).
The consequence of glycolytic metabolism in any tissue is the formation of H+, which must be removed from the cell if the internal milieu is to maintain its normal pH, because many cellular processes have a narrow pH optimum. Four major types of intracellular pH (pHi) regulatory mechanisms have been identified in tumour cells: Na+/H+ exchangers, bicarbonate transporters, proton-lactate symporters and proton pumps (Martinez-Zaguilan et al. Citation1993, Robertson et al. Citation2004, Sennoune et al. Citation2004). These transmembrane proteins are ion pumps or ion exchangers that pump protons across the plasma membrane from the cytoplasm to the opposite site of the membrane, the extracellular space or the lumen of various organelles. A consequence of the activity of ion pumps is an enhanced pH gradient across the plasma membrane of cancer cells in comparison with normal cells, and a lower pH in the extracellular milieu (Tannock and Rotin Citation1989, Raghunand et al. Citation1999a, Gatenby and Gillies Citation2004, Becelli et al. Citation2007).
Usually, exposure to an acidic environment results in cell death, however cancer cells adapt through resistance to apoptosis and up-regulation of membrane ion channels in order to maintain intracellular pH in the range of normality (Gatenby and Gillies Citation2004). Indeed, the unfavorable environment may favor tumour cell survival in acidic conditions via selection of cells that are resistant to acid-induced cell toxicity and hypoxia-induced, p53-dependent apoptosis (Graeber et al. Citation1996, Park et al. Citation1999, Williams et al. Citation1999, Gatenby et al. Citation2007), and promote invasiveness by killing normal tissue cells. Malignant tumour cells not only survive better in acidic environments, but they also demonstrate phagocytotic and cannibalistic behavior (Lugini et al. Citation2006, Fais Citation2007). Extracellular acidification promotes cancer invasion and metastasis by increased secretion and activation of proteases, matrix metalloproteinases, bone morphogenetic protein-1-type metalloproteinases, tissue serine proteases, and adamalysin-related membrane proteases (Martinez-Zaguilan et al. Citation1996, Bhujwalla et al. Citation2002, Rofstad et al. Citation2006). Enhanced mutation rates (Yuan and Glazer Citation1998, Bindra and Glazer Citation2005), chromosomal instability (Morita et al. Citation1991), and spontaneous transformation (LeBoeuf et al. Citation1990) are associated with acidity. Hypoxia and acidity also cause resistance to radiotherapies and chemotherapies (Vaupel et al. Citation1989, Raghunand et al. Citation1999c, Raghunand and Gillies Citation2001), and promote the expression of the human multi-drug-resistance protein (Wei and Roepe Citation1994).
Tumour acidity might be an alternative to specific molecular biomarkers for tumour targeting and detection and may also be useful for monitoring therapy outcomes. Recently it has been shown that the level of extracellular pH is related to the overall survival of canines with spontaneous sarcomas. Thus, the pH was predictive of a clinical outcome (Lora-Michiels et al. Citation2006). The advantages of targeting acidity include its generality and the absence of tumour heterogeneity issues.
The need for hydrophobicity has been a constraint on drugs
If the target of a therapeutic is cytoplasmic, the selective delivery of therapeutics to a tumour is not enough to improve treatment; the strategy must also enable the agent to cross the hydrophobic barrier of a cell membrane to act inside cells. The two major mechanisms for the translocation of molecules and nanoparticles across the membrane are passive diffusion and endocytosis, although the latter still relies on permeation of the endosomal membrane (Andresen et al. Citation2005, Kim Citation2007). Neither is specific for cancer cells, so each would promote translocation of therapeutics across the membranes of cells in both diseased and healthy tissues. In conventional drug design and discovery the Lipinski rules of five are widely used to guide molecular designs. The rules postulate that a successful drug should be hydrophobic and small in order to traverse membranes and reach cytoplasmic targets (e.g., the logarithm of the octanol-water partition coefficient LogPo/w is −0.4 to +5.6 and the MW is 160–480 g.mol−1) (Lipinski et al. Citation2001). Drugs designed in this way will indiscriminately enter all cells they encounter, and are also likely to be substrates for efflux pumps that reduce their efficacy. It is important to note that the majority of inhibitors found for biological targets located inside a cell are molecules (in most cases, peptides) that cannot cross a membrane (Wang et al. Citation2000, Tsutsumi and Neckers Citation2007, Sun et al. Citation2008). Another large class of cell-impermeable functional molecules comprises gene regulation agents such as DNA, siRNA, and PNA (peptide nucleic acid) (Knudsen and Nielsen Citation1997, Elayadi and Corey Citation2001). Gene-targeted therapies might lead to a revolution in cancer therapeutics; however, the delivery of these agents, involving passage through the cell membrane, appears to be a general problem (Nielsen Citation2005).
Cell-penetrating peptides are used as one approach to the delivery of liposomes, nanoparticles, adenoviruses, and a variety of biological molecules into cells (Thoren et al. Citation2000, Munoz-Morris et al. Citation2007, Patel et al. Citation2007). Among these peptides are TAT (peptide derived from the trans-activating transcriptional activator (TAT) protein), antennapedia, arginine-rich peptides and others. The mechanisms of entry are still under discussion (Vives et al. Citation2003) but it is most likely they enter cell via the endocytic pathway and act cooperatively. When taken up by endocytosis, molecules or nanoparticles are trapped in the lysosome compartment and need to be released into the cytoplasm. To induce this process, a pH-sensitive polymer coating has been developed (Ganta et al. Citation2008). Usually, the polymers contain multiple carboxyl groups, which become protonated in an acidic endosome (pH 5.0) and change their properties to break through the membrane.
Membrane peptides as a novel class of delivery agents
The folding and insertion into membrane of constitutive membrane proteins is facilitated by complex molecular machines in vivo, including the translocon that places most transmembrane helices across the bilayer (Van den Berg et al. Citation2004, White and von Heijne Citation2004, Osborne et al. Citation2005). At the same time, non-constitutive membrane proteins, such as toxins, antimicrobial peptides, C-tail proteins insert themselves into a lipid bilayer without assistance. It has been found that moderately polar transmembrane domains with sequences of up to 85 residues can translocate themselves into membranes in a translocon-defective yeast strain, in contrast to more hydrophobic sequences that cannot (Brambillasca et al. Citation2005, Citation2006). Further, there is a class of synthetic peptides that fold and insert when they interact with a lipid bilayer (Wimley and White Citation2000, Holt and Killian Citation2009). Because the spontaneous insertion and folding of a peptide into a lipid bilayer should seek the free energy minimum, an insertion event is accompanied by a release of energy (Engelman et al. Citation2003). Therefore, it is reasonable to suppose that the energy might be used to translocate cell-impermeable cargo molecules across a cellular membrane. It follows that moderately hydrophobic peptides might be considered as the basis for a novel class of delivery agents. However, several key properties must be preserved for practical use, including aqueous solubility and lack of toxicity.
pH (Low) Insertion Peptide: pHLIP
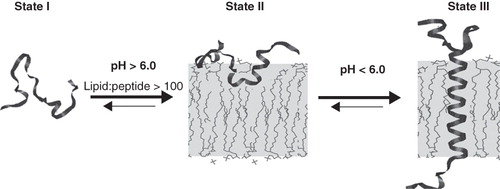
Attempts have been made to design synthetic peptides that are soluble in aqueous solution, and that spontaneously and selectively insert into a membrane (Wimley and White Citation2000). The only significant success to date remains the discovery of pHLIP, named from: pH (Low) Insertion Peptide (). pHLIP is a water-soluble polypeptide derived from the bacteriorhodopsin C helix, which was found to insert across a membrane to form a stable transmembrane alpha helix (Hunt et al. Citation1997). Peptide folding and membrane insertion are driven by a drop of pH from neutral or high (>7.4) to slightly acidic (7.0–6.5 and less) pHs. The apparent pK of insertion was found to be 6.0 (Hunt et al. Citation1997, Reshetnyak et al. Citation2008). pHLIP is a monomer in each of its three major states: unstructured and soluble in water (state I) at neutral pH, unstructured and bound to the surface of a membrane at neutral pH (state II), and inserted across the membrane as an α-helix at low pH (state III) ().
Figure 1. The sequence of pHLIP peptide.

Figure 2. The three major states of pHLIP at a concentration of < 30 μg/ml are illustrated: Unstructured and soluble in water at pH > 7 (state I); unstructured and bound to the surface of a lipid bilayer at the same pH and at a lipid:peptide molar ratio >100 (state II); and inserted across the bilayer as an α-helix at low pH (state III). The Figure is taken from Reshetnyak et al. (Citation2007).
Toxicity
Toxicity is one of the most critical issues in the selection of any delivery agent. For example, the use of pore-forming membrane peptides as delivery agents is complicated by the toxicity associated with the formation of pores in cellular membranes in vivo (Shai Citation1999). By contrast, the interaction of pHLIP with liposomes and cellular membranes at both neutral and low pHs does not lead to membrane leakage (Reshetnyak et al. Citation2007, Zoonens et al. Citation2008), and no cellular toxicity was seen over a range of peptide concentrations (Andreev et al. Citation2007). Also, mice receiving a high dose (about 5 mg/kg) of peptide did not show any obvious adverse effects within two months after intravenous peptide administration (Andreev et al. Citation2009). Despite these encouraging observations, a systematic toxicity study is needed.
Selectivity of targeting
The pH-dependent interaction of pHLIP with membranes allows selectivity in the targeting of acidic diseased tissue. As noted above, acidity and hypoxia are considered as universal cancer biomarkers, and pHLIP is used as an acidity-targeting probe (Andreev et al. Citation2009, Vavere et al. Citation2009). Besides cancer, many other pathological states, such as inflammation, ischemia, stroke, arthritis and others are characterized by acidity in the extracellular space, which may broaden the potential applications of pHLIP (Kellum et al. Citation2004, Xiong et al. Citation2008, Holzer Citation2009). In vivo fluorescence imaging in mice and rats demonstrates that pHLIP can target acidic tissues, such as kidneys, tumours of various sizes and origins, and the site of experimentally induced inflammatory arthritis (Andreev et al. Citation2009). In addition to fluorescence imaging, PET (positron emission tomography) imaging of the acidic environment in human prostate tumours was performed using 64Cu-DOTA conjugated to pHLIP (Vavere et al. Citation2009). PET studies demonstrated that the construct avidly accumulated in LNCaP and PC-3 tumours and that tumour uptake correlates with the differences in the bulk extracellular pH (pHe) measured by MR spectroscopy. Feeding animals with bicarbonated water, which increases tissue pH (Raghunand et al. Citation1999b), results in a reduction of tumour targeting by pHLIP (Vavere et al. Citation2009).
Molecular mechanism of pH-dependent membrane insertion of pHLIP
The putative transmembrane (TM) part of pHLIP peptide contains two Asp residues (see ). At neutral pH these charged residues enhance peptide solubility and serve as anchors keeping the peptide at the surface of membrane, thereby preventing pHLIP partitioning into the hydrophobic membrane bilayer (Andreev et al. Citation2009, Musial-Siwek et al. Citation2010). A reduction of pH induces protonation of Asp residues, and as a result, the overall hydrophobicity of the peptide increases, enhancing the affinity of the peptide for the lipid bilayer core and triggering peptide folding and insertion. The replacement of the key Asp residues by Lys, Ala or Asn leads to the loss of peptide of pH-dependent membrane insertion, as measured in liposomes, red blood cells and confirmed by in vivo fluorescence imaging (Andreev et al. Citation2009, Musial-Siwek et al. Citation2010). The K-pHLIP peptide, where the two Asp residues in the putative transmembrane region are replaced with Lys residues, does not demonstrate tumour targeting (Andreev et al. Citation2009, Musial-Siwek et al. Citation2010). The Ala substitutions give a peptide that aggregates in solution, while the Lys and Asn substitutions give peptides that are too polar to insert either at neutral or low pH. The replacement of one of the Asp residues in the TM part of the peptide by a Glu residue results in a shift of pH of membrane insertion from 6.0 to 6.5 (Andreev et al. Citation2009, Musial-Siwek et al. Citation2010). Replacement of both Asp residues by Glu results in enhancement of peptide aggregation and formation of elements of secondary structure on the bilayer surface at neutral pH. There are experimental data, as well as theoretical calculations indicating that pK of protonation/deprotonation of various amino acids located at border of lipid bilayer or inside membrane could be significantly altered (Balashov Citation2000, Ladokhin and White Citation2004, Yoo and Cui Citation2008, Ghosh et al. Citation2009). Therefore it is not surprising to observe that the apparent pK for Asp residues in pHLIP is 6.0 and could be altered to higher or lower values.
Data obtained on model systems (liposomes), cultured cells and mice allow us to conclude that the mechanism of membrane entry of pHLIP is not mediated by endocytosis, interactions with cell receptors or pore formation (Reshetnyak et al. Citation2006, Reshetnyak et al. Citation2007, Andreev et al. Citation2009); rather, the mechanism is simply the formation of a helix across the lipid bilayer, triggered by the increase of peptide hydrophobicity due to the protonation of negatively charged residues induced by low pH.
Solubility and stability of pHLIP in blood
Poor solubility due to aggregation is a typical property of membrane peptides, which has complicated studies and applications. pHLIP, as any membrane peptide, also has a tendency to aggregate, especially at high concentrations and/or low pH (Reshetnyak et al. Citation2007, Musial-Siwek et al. Citation2010). However, in aqueous solution at neutral pH pHLIP exists as a monomer at concentrations less than 30 μg/ml (∼ 7.0 μM), as studied by fluorescence and CD spectroscopy measurements, size exclusion chromatography coupled with ‘on-line’ laser light scattering, ultraviolet and refractive index detection (SEC-LS/UV/RI) and analytical ultracentrifugation experiments (Reshetnyak et al. Citation2007). Our studies indicate that, when the solubility of the peptide is compromised as a result of mutations, the affinity of the peptide for a membrane and its overall conformational properties can change.
The oligomeric state of the peptide on the surface of a membrane (state II) and inserted into the lipid bilayer (state III) were evaluated by FRET performed with two different donor-acceptor probes attached to the N-terminus of the peptide (Reshetnyak et al. Citation2007). The data demonstrate that, at low concentrations, the peptide is monomeric in both states II and III.
Peptide interactions with proteins, especially plasma proteins, and membranes determine the pharmacokinetics of the peptide at neutral pH. pHLIP demonstrates prolonged circulation in the blood (several hours), which is consistent with its ability to bind weakly to membrane surfaces at neutral and high pH, preventing the rapid clearance by the kidney expected for a small, soluble peptide (Reshetnyak et al. Citation2008, Andreev et al. Citation2009). According to our data, pHLIP binding to membranes is mostly driven by hydrophobic interactions (Reshetnyak et al. Citation2008). If the peptide sequence were made more hydrophobic, we would expect tighter binding to red blood cells and epithelial cells and more aggregation in solution, and slower clearance and reduced bioavailability. On the other hand, making the peptide less hydrophobic might accelerate clearance and prevent the peptide from finding its targets. Therefore, solubility is an important property to manage as applications in vivo are developed.
Another important property is the stability of peptides in the blood, since proteases in the serum can degrade peptides consisting of L-amino acids within minutes. While polypeptides made from D-amino acids are much more stable (Hong et al. Citation1999), they are often unsuitable for specific receptor binding applications as a consequence of their altered chirality. Since the mechanism of pHLIP involves relatively non-specific interactions with a fluid lipid bilayer, it is not a surprise that pHLIP peptides composed of L- or D-amino acids demonstrate the same biophysical and tumour-targeting properties (Andreev et al. Citation2009). This observation adds to the evidence that the pHLIP targeting does not require any specific molecular binding event. The only conspicuous difference is that D-pHLIPs form left-handed helices across membranes rather than the right-handed helices formed by L-pHLIPs (Andreev et al. Citation2009).
Topology of membrane insertion
Is there a specific orientation of pHLIP insertion, or can either end insert? The topology of pHLIP insertion was probed using an NBD-dithionite quenching assay (McIntyre and Sleight Citation1991), and then confirmed in experiments on cultured cells (Reshetnyak et al. Citation2006, Thevenin et al. Citation2009). NDB and IANBD were covalently attached to the N- and the C-terminus of pHLIP peptides, respectively (Reshetnyak et al. Citation2006, Citation2007). Fluorescently labeled peptides were inserted into the lipid bilayer at low pH, and changes in the fluorescence signal of NBD were measured after quenching by dithionite, a membrane-impermeable agent that abolishes NBD fluorescence if it contacts the dye. The data clearly indicate that the N-terminus of pHLIP stays outside of the bilayer, while the C-terminus inserts across the lipid bilayer at low pH (Reshetnyak et al. Citation2006, Citation2007).
Cargo translocation by pHLIP insertion
What is the source and magnitude of the insertion energy that might be used to move cargo across a membrane? The partition of pHLIP into the outer leaflet of lipid bilayer at neutral pH and the folding/insertion at low pH are accompanied by the release of energy (Reshetnyak et al. Citation2008). Fluorescence spectroscopy, isothermal titration calorimetry and acid titration calorimetry were used to study the interactions of pHLIP with a POPC lipid bilayer and to calculate the transition energies between states. The Gibbs Free Energy of binding to a POPC surface (state I – state II transition) at 37°C is about −7 kcal/mol near neutral pH and the additional free energy of insertion and folding across a lipid bilayer at low pH (state II – state III transition) is nearly −2 kcal/mol (Reshetnyak et al. Citation2008). The energy difference between state II and state III could be used to favour the partition of cargo across the hydrophobic bilayer of membrane.
To experimentally evaluate the range of cargo polarity that pHLIP can move across a membrane we used an assay based on the NDB-dithionite quenching experiments described above (Thevenin et al. Citation2009). NDB was attached to cyclic peptides having a range of hydrophobicity, with calculated LogPo/w values at low pH ranging from −2.6 to −8.5. The cyclic peptides were conjugated to a Cys residue at the C-terminus of pHLIP via an S-S bond, and translocation was accessed by quenching of NBD fluorescence. We found that cargo molecules with a LogPo/w of ∼−2.6 and MW of ∼800 are delivered into both liposomes and cancer cells at low pH with an efficiency of about 70%, while cargo molecules with calculated LogPo/w values of −8 could not be moved across either membrane (Thevenin et al. Citation2009).
Kinetics of pHLIP insertion into membrane
While the equilibrium thermodynamics favor binding and insertion of pHLIP, slow kinetics could be limiting for in vivo use, since blood flow is very fast. Kinetic studies of pHLIP folding and insertion across a POPC lipid bilayer triggered by a pH drop from 8.0 to 4.0 indicate that insertion takes 100 sec, with a rapid (0.1 sec) interfacial helix formation followed by insertion to give a transmembrane helix (Andreev et al. Citation2010). In the case of a pH drop to 6.0 the insertion is slower, about 300 sec (unpublished work). However, recent data obtained on various pHLIP variants show that the process of insertion can be accelerated by 10–100 times if acidic residues are removed from the C-terminus of the peptide (unpublished work).
Dual delivery capability of pHLIP
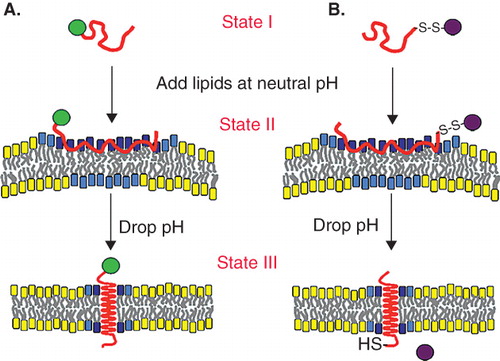
pHLIP, in contrast to cell-penetrating peptides, stays in the cellular membrane after insertion, translocating one end into cytoplasm and leaving the other end in the extracellular space (Andreev et al. Citation2009). Therefore, the peptide possesses dual delivery capabilities: it can tether cargo molecules to the cell surface and/or it can inject and release cell-impermeable cargo molecules into the cytoplasm (). In the first scenario, a cargo molecule is attached to the pHLIP N-terminus. Suitable external cargo molecules might possess a wide range of polarity and size. One of the applications can be to deliver imaging probes to acidic tissue, where they will be stably tethered to the surfaces of cells. Our recent data indicate that pHLIP can deliver and tether various nanoparticles to the surface of cancer cells (unpublished data). The second delivery capability of pHLIP is based on the conjugation of cargo molecules to the C-terminus via a bond that is cleaved in the environment of the cytoplasm, such as a disulfide. Since the energy released during peptide folding and insertion across a membrane is limited, and since strongly polar molecules will reach equilibrium slowly, there is a limit on cargo polarity (and most probably on size as well) that can be delivered across a membrane by pHLIP, even taking into account the mass action effect discussed above. For example, we did not succeed in the translocation of a 20 base oligonucleotide, probably because of its highly charged backbone (Reshetnyak et al. Citation2006).
Figure 3. A schematic representation of the dual delivery capabilities of pHLIP is shown: (A) Tethering of cargo molecules to the surface of cells with low extracellular pH, and (B) translocation of cell-impermeable polar cargo molecules across the membrane lipid bilayer. State I corresponds to the peptide in solution at normal and basic pHs. By addition of vesicles, the unstructured peptide is adsorbed on the membrane surface, raising the local concentration (State II). A drop of pH leads to the protonation of Asp residues, increasing peptide hydrophobicity, and resulting in the insertion and formation of a transmembrane alpha-helix (State III). Lipids interacting with the peptide directly are marked with blue head groups, lipids influenced by the interaction but not interacting with the peptide directly have cyan head groups, and lipids that are not involved in the interaction with pHLIP have yellow head groups. The Figure is reprinted from Andreev et al. (Citation2009) (http://chemistry-today.teknoscienze.com/).
pHLIP-mediated translocation of cell-impermeable functional cargos
As already discussed, pHLIP can translocate cargo molecules attached to its C-terminus. Translocation is selective for low pH, and various types of cargo molecules attached by disulfides can be released in the cytoplasm, including various fluorescent dyes, synthetic cyclic peptides, toxins and peptide nucleic acids (Reshetnyak et al. Citation2006). We have shown that a cell-impermeable fluorescently-labelled toxin, phalloidin-rhodamine, conjugated to the C-terminus of pHLIP via an S-S- bond, can be moved across the membrane in a pH-dependent manner (Reshetnyak et al. Citation2006) and unpublished work. The pH-dependent translocation of the fluorescent phalloidin by the peptide was confirmed by fluorescence microscopy and fluorescence activated cell sorting. If phalloidin-rhodamine enters a cell, it binds tightly to actin filaments at nanomolar concentration (KD = 40 nM) and strongly inhibits their depolymerization (Wehland et al. Citation1977). Actin filaments stained with fluorescent phalloidin have an unmistakable filamentous pattern, distinct from the appearance of other cellular structures, organelles or membrane staining. The phalloidin translocated into the cytoplasm of live cells inhibits the proliferation, contractility, migration and division of cells. A long term effect of phalloidin is the formation of multinucleated cells, since nuclei can divide in treated cells, but the cell itself cannot. This process leads to the formation of multiple nuclei in one cell and eventual cell death.
Another example is the translocation of a class of cell-impermeable functional cargo-molecule, peptide nucleic acids (PNA), by pHLIP (Reshetnyak et al. Citation2006). PNAs can base pair specifically to target nucleic acid sequences, but lack the highly charged backbone of biological nucleic acids, and are therefore candidates for pHLIP delivery. In vitro studies show that PNA can inhibit both transcription and translation of genes to which it has been targeted, which suggests use of PNA in antigene and antisense therapy (Nielsen Citation2005). However, a major obstacle has been the delivery of PNA (as well as RNA or ODN) across membranes into cells. We have demonstrated the ability of pHLIP to translocate a fluorescence-labeled 12 base PNA into cells (Reshetnyak et al. Citation2006). Treatment of cells with PNA-rhodamine alone did not give fluorescent staining of cells at pH 6.5 or 7.4, but fluorescence was observed in cells when the PNA was linked to the C-terminus of pHLIP via a disulfide and added at pH 6.5. We verified that the labelled cells were alive by using the dead cell marker SYTOX-Green. In more recent in vivo studies we find that pHLIP can deliver an18-base PNA (MW 4.7 kDa) to a mouse tumour, translocate PNA across membranes, and activate luciferase expression in a result of splicing correction (unpublished work).
pHLIPs are a new class of delivery agents
In summary, we would like to outline the general features that define the pHLIP class of polypeptides. To date, we have studied more than 20 different variants of the parent pHLIP sequence given in . The sequence is presented as having three main blocks. The middle part is a moderately polar sequence that contains protonatable residues, and is the environment-sensitive, membrane inserting part of the peptide. Recent studies show that the transmembrane helix might be as short as 15 residues (London and Shahidullah Citation2009), therefore we assume that the membrane-inserting sequences might consist of ∼15 to 25 amino acids. The other two blocks are the two flanking sequences. The role of the flanking sequences is mostly to modulate the peptide solubility, but might also include functional motifs; for example, protease cleavage or receptor binding sequences, or amino acids that can undergo phosphorylation in the cytoplasm. There are several restrictions applied to the flanking sequence that inserts across the membrane: (i) It should not contain many charged residues (especially positive charges); (ii) it should not be long; and (iii) the speed and cooperativity of peptide insertion into membrane will depend on the number of Asp or Glu residues present in this sequence. On the other hand, there is no specific restriction to the flanking sequence that stays outside of membrane other than its role in peptide solubility; however a danger is that dramatic changes of peptide pharmacokinetics might result from extreme variations. The membrane-inserting sequence of pHLIP does not contain Cys or Lys residues. Any of these residues or both could be placed at the N- or C-terminus of the peptide for the purpose of cargo conjugation to pHLIP (NHS and maleimide click chemistry is developed very well and widely used for conjugation purposes). We are optimistic that the pHLIP technology will have utility in a variety of medical and scientific applications.
Acknowledgements
We thank all the members of the Andreev, Engelman and Reshetnyak laboratories for their essential contributions to the pHLIP project.
Declaration of interest: This work was supported in part by grants from the Department of Defence BCRP CDMRP BC061356 and the National Institutes of Health National Cancer Institute RCA125280A to OAA, RO1133890 to OAA, DME, YKR, and GM073857 to DME.
References
- Aghi M, Martuza RL. 2005. Oncolytic viral therapies – the clinical experience. Oncogene, 24:7802–7816.
- Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, Engelman DM, Reshetnyak YK. 2007. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc Natl Acad Sci USA 104:7893–7898.
- Andreev OA, Engelman DM, Reshetnyak YK. 2009. Targeting acidic diseased tissue. New technology based on use of the pH (Low) Insertion Peptide (pHLIP). Chemistry Today 27:10–13.
- Andreev OA, Karabadzhak AG, Weerakkody D, Andreev GO, Engelman DM, Reshetnyak YK. 2010. The pHLIP peptide inserts across a lipid bilayer as a helix, and exits by a different path Proc Natl Acad Sci USA.
- Andresen TL, Jensen SS, Jorgensen K. 2005. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog Lipid Res 44:68–97.
- Balashov SP. 2000. Protonation reactions and their coupling in bacteriorhodopsin. Biochim Biophys Acta 1460:75–94.
- Becelli R, Renzi G, Morello R, Altieri F. 2007. Intracellular and extracellular tumor pH measurement in a series of patients with oral cancer. J Craniofac Surg 18:1051–1054.
- Bhujwalla ZM, Artemov D, Ballesteros P, Cerdan S, Gillies RJ, Solaiyappan M. 2002. Combined vascular and extracellular pH imaging of solid tumors. NMR Biomed 15:114–119.
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA Jr, Marks JR, Dressman HK, West M, Nevins JR. 2006. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439:353–357.
- Bindra RS, Glazer PM. 2005. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat Res 569:75–85.
- Blättler WA, Chari RVJ. 2001. Drugs to enhance the therapeutic potency of anticancer antibodies, antibody-drug conjugates as tumor-activated prodrugs. Washington, DC: American Chemical Society.
- Brambillasca S, Yabal M, Makarow M, Borgese N. 2006. Unassisted translocation of large polypeptide domains across phospholipid bilayers. J Cell Biol 175:767–777.
- Brambillasca S, Yabal M, Soffientini P, Stefanovic S, Makarow M, Hegde RS, Borgese N. 2005. Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J 24:2533–2542.
- Buchsbaum DJ. 2004. Imaging and therapy of tumors induced to express somatostatin receptor by gene transfer using radiolabeled peptides and single chain antibody constructs. Semin Nucl Med 34:32–46.
- Carter P. 2001. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 1:118–129.
- Davis ME, Chen ZG, Shin DM. 2008. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat Rev Drug Discov 7:771–782.
- de Bono JS, Tolcher AW, Forero A, Vanhove GF, Takimoto C, Bauer RJ, Hammond LA, Patnaik A, White ML, Shen S, Khazaeli MB, Rowinsky EK, LoBuglio AF. 2004. ING-1, a monoclonal antibody targeting Ep-CAM in patients with advanced adenocarcinomas. Clin Cancer Res 10:7555–7465.
- Duguet E, Vasseur S, Mornet S, Devoisselle JM. 2006. Magnetic nanoparticles and their applications in medicine. Nanomedicine 1:157–168.
- Elayadi AN, Corey DR. 2001. Application of PNA and LNA oligomers to chemotherapy. Curr Opin Investig Drugs 2:558–561.
- Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. 2003. Membrane protein folding: Beyond the two stage model. FEBS Lett 555:122–125.
- Everts B, van der Poel HG. 2005. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther 12:141–161.
- Fais S. 2007. Cannibalism: A way to feed on metastatic tumors. Cancer Lett 258:155–164.
- Freimark B, Clark D, Pernasetti F, Nickel J, Myszka D, Baeuerle PA, Van Epps D. 2007. Targeting of humanized antibody D93 to sites of angiogenesis and tumor growth by binding to multiple epitopes on denatured collagens. Mol Immunol 44:3741–3750.
- Ganta S, Devalapally H, Shahiwala A, Amiji M. 2008. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release 126:187–204.
- Gatenby RA, Gillies RJ. 2004. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899.
- Gatenby RA, Gillies RJ. 2008. A microenvironmental model of carcinogenesis. Nat Rev Cancer 8:56–61.
- Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. 2007. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer 97:646–653.
- Ghosh N, Prat-Resina X, Gunner MR, Cui Q. 2009. Microscopic pKa analysis of Glu286 in cytochrome c oxidase (Rhodobacter sphaeroides): Toward a calibrated molecular model. Biochemistry 48:2468–2485.
- Gillies RJ, Liu Z, Bhujwalla Z. 1994. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am J Physiol 267:C195–203.
- Gillies RJ, Robey I, Gatenby RA. 2008. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med 49(Suppl. 2):24S–42S.
- Gillies RJ, Schornack PA, Secomb TW, Raghunand N. 1999. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1:197–207.
- Gindy ME, Prud'homme RK. 2009. Multifunctional nanoparticles for imaging, delivery and targeting in cancer therapy. Expert Opin Drug Deliv 6:865–878.
- Goldsmith SJ. 1997. Receptor imaging: Competitive or complementary to antibody imaging? Semin Nucl Med 27:85–93.
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. 1996. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379:88–91.
- Griffiths JR. 1991. Are cancer cells acidic? Br J Cancer 64:425–427.
- Hainfeld JF, Slatkin DN, Smilowitz HM. 2004. The use of gold nanoparticles to enhance radiotherapy in mice. Phys Med Biol 49:309–315.
- Hanke JH, Webster KR, Ronco LV. 2004. Protein biomarkers and drug design for cancer treatments. Eur J Cancer Prev 13:297–305.
- Holt A, Killian JA. 2009. Orientation and dynamics of transmembrane peptides: The power of simple models. Eur Biophys J 107:4081–4086.
- Holzer P. 2009. Acid-sensitive ion channels and receptors. Handb Exp Pharmacol 283–332.
- Hong SY, Oh JE, Lee KH. 1999. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem Pharmacol 58:1775–1780.
- Hunt JF, Rath P, Rothschild KJ, Engelman DM. 1997. Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry 36:15177–15192.
- Ito A, Shinkai M, Honda H, Kobayashi T. 2005. Medical application of functionalized magnetic nanoparticles. J Biosci Bioeng 100:1–11.
- Janssens JP, Verlinden I, Gungor N, Raus J, Michiels L. 2004. Protein biomarkers for breast cancer prevention. Eur J Cancer Prev 13:307–317.
- Jeffrey SS, Lonning PE, Hillner BE. 2005. Genomics-based prognosis and therapeutic prediction in breast cancer. J Natl Compr Canc Netw 3:291–300.
- Kankaanranta L, Seppala T, Koivunoro H, Saarilahti K, Atula T, Collan J, Salli E, Kortesniemi M, Uusi-Simola J, Makitie A, Seppanen M, Minn H, Kotiluoto P, Auterinen I, Savolainen S, Kouri M, Joensuu H. 2007. Boron neutron capture therapy in the treatment of locally recurred head and neck cancer. Int J Radiat Oncol Biol Phys 69:475–482.
- Kellum JA, Song M, Li J. 2004. Science review. Extracellular acidosis and the immune response: Clinical and physiologic implications. Crit Care 8:331–336.
- Kim KY. 2007. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomedicine 3:103–110.
- Knudsen H, Nielsen PE. 1997. Application of peptide nucleic acid in cancer therapy. Anticancer Drugs 8:113–118.
- Krebs HA. 1972. The Pasteur effect and the relations between respiration and fermentation. Essays Biochem 8:1–34.
- Krenning EP, de Jong M, Kooij PP, Breeman WA, Bakker WH, de Herder WW, van Eijck CH, Kwekkeboom DJ, Jamar F, Pauwels S, Valkema R. 1999. Radiolabelled somatostatin analogue(s) for peptide receptor scintigraphy and radionuclide therapy. Ann Oncol 10(Suppl. 2):S23–29.
- Ladokhin AS, White SH. 2004. Interfacial folding and membrane insertion of a designed helical peptide. Biochemistry 43:5782–5791.
- Latorre M, Rinaldi C. 2009. Applications of magnetic nanoparticles in medicine: Magnetic fluid hyperthermia. P R Health Sci J 28:227–238.
- LeBoeuf RA, Kerckaert GA, Aardema MJ, Gibson DP. 1990. Multistage neoplastic transformation of Syrian hamster embryo cells cultured at pH 6.70. Cancer Res 50:3722–3729.
- Liapi E, Geschwind JF. 2007. Transcatheter and ablative therapeutic approaches for solid malignancies. J Clin Oncol 25:978–986.
- Lin E, Nemunaitis J. 2004. Oncolytic viral therapies. Cancer Gene Ther 11:643–664.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26.
- Lister-James J, Moyer BR, Dean RT. 1997. Pharmacokinetic considerations in the development of peptide-based imaging agents. Q J Nucl Med 41:111–118.
- London E, Shahidullah K. 2009. Transmembrane vs. non-transmembrane hydrophobic helix topography in model and natural membranes. Curr Opin Struct Biol 19:464–472.
- Lora-Michiels M, Yu D, Sanders L, Poulson JM, Azuma C, Case B, Vujaskovic Z, Thrall DE, Charles HC, Dewhirst MW. 2006. Extracellular pH and P-31 magnetic resonance spectroscopic variables are related to outcome in canine soft tissue sarcomas treated with thermoradiotherapy. Clin Cancer Res 12:5733–5740.
- Lu H, Goodell V, Disis ML. 2007. Targeting serum antibody for cancer diagnosis: A focus on colorectal cancer. Expert Opin Ther Targets 11:235–244.
- Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, Gentile M, Luciani F, Parmiani G, Rivoltini L, Malorni W, Fais S. 2006. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res 66:3629–3638.
- Ma L, Yu P, Veerendra B, Rold TL, Retzloff L, Prasanphanich A, Sieckman G, Hoffman TJ, Volkert WA, Smith CJ. 2007. In vitro and in vivo evaluation of Alexa Fluor 680-bombesin[7–14]NH2 peptide conjugate, a high-affinity fluorescent probe with high selectivity for the gastrin-releasing peptide receptor. Mol Imaging 6:171–180.
- Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. 1993. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol 265:C1015–1029.
- Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. 1996. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis 14:176–186.
- McIntyre JC, Sleight RG. 1991. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30:11819–11827.
- Mitin VN, Kulakov VN, Khokhlov VF, Sheino IN, Arnopolskaya AM, Kozlovskaya NG, Zaitsev KN, Portnov AA. 2009. Comparison of BNCT and GdNCT efficacy in treatment of canine cancer. Appl Radiat Isot 67:S299–301.
- Moasser MM. 2007. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 26:6577–6592.
- Moghimi SM. 2006. Recent developments in polymeric nanoparticle engineering and their applications in experimental and clinical oncology. Anticancer Agents Med Chem 6:553–561.
- Morita T, Nagaki T, Fukuda I, Okumura K. 1991. Effect of pH on the activity and stability of clastogens in the in vitro chromosomal aberration test with Chinese hamster ovary K1 cells. Mutat Res 262:159–166.
- Morris KN, Weil MD, Malzbender R. 2006. Radiochromic film dosimetry of contrast-enhanced radiotherapy (CERT). Phys Med Biol 51:5915–5925.
- Munoz-Morris MA, Heitz F, Divita G, Morris MC. 2007. The peptide carrier Pep-1 forms biologically efficient nanoparticle complexes. Biochem Biophys Res Commun 355:877–882.
- Musial-Siwek M, Karabadzhak A, Andreev OA, Reshetnyak YK, Engelman DM. 2010. Tuning the insertion properties of pHLIP. Biochim Biophys Acta 1798:1041–1046.
- Muxi A, Pons F, Vidal-Sicart S, Setoain FJ, Herranz R, Novell F, Fernandez RM, Trias M, Setoain J. 1999. Radioimmunoguided surgery of colorectal carcinoma with an 111In-labelled anti-TAG72 monoclonal antibody. Nucl Med Commun 20:123–130.
- Newell K, Franchi A, Pouyssegur J, Tannock I. 1993. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci USA 90:1127–1131.
- Nielsen PE. 2005. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA). Q Rev Biophys 38:345–350.
- O'Connell MJ, Bachilo SM, Huffman CB, Moore VC, Strano MS, Haroz EH, Rialon KL, Boul PJ, Noon WH, Kittrell C, Ma J, Hauge RH, Weisman RB, Smalley RE. 2002. Band gap fluorescence from individual single-walled carbon nanotubes. Science 297:593–596.
- O'Connor AE, Gallagher WM, Byrne AT. 2009. Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy. Photochem Photobiol 85:1053–1074.
- Osborne AR, Rapoport TA, van den Berg B. 2005. Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol 21:529–550.
- Palumbo G. 2007. Photodynamic therapy and cancer: A brief sightseeing tour. Expert Opin Drug Deliv 4:131–148.
- Park HJ, Lyons JC, Ohtsubo T, Song CW. 1999. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer 80:1892–1897.
- Patel LN, Zaro JL, Shen WC. 2007. Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives. Pharm Res 24:1977–1992.
- Payne G. 2003. Progress in immunoconjugate cancer therapeutics. Cancer Cell 3:207–212.
- Pisarev MA, Dagrosa MA, Juvenal GJ. 2007. Boron neutron capture therapy in cancer: Past, present and future. Arq Bras Endocrinol Metabol 51:852–856.
- Polascik TJ, Manyak MJ, Haseman MK, Gurganus RT, Rogers B, Maguire RT, Partin AW. 1999. Comparison of clinical staging algorithms and 111indium-capromab pendetide immunoscintigraphy in the prediction of lymph node involvement in high risk prostate carcinoma patients. Cancer 85:1586–1592.
- Raghunand N, Altbach MI, van Sluis R, Baggett B, Taylor CW, Bhujwalla ZM, Gillies RJ. 1999a. Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by 31P magnetic resonance spectroscopy. Biochem Pharmacol 57:309–312.
- Raghunand N, Gillies RJ. 2001. pH and chemotherapy. Novartis Found Symp 240:199–211; discussion 265–268.
- Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D, Bhujwalla ZM, Gillies RJ. 1999b. Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer 80:1005–1011.
- Raghunand N, Martinez-Zaguilan R, Wright SH, Gillies RJ. 1999c. pH and drug resistance. II. Turnover of acidic vesicles and resistance to weakly basic chemotherapeutic drugs. Biochem Pharmacol 57:1047–1058.
- Reshetnyak YK, Andreev OA, Lehnert U, Engelman DM. 2006. Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. Proc Natl Acad Sci USA 103:6460–6465.
- Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. 2008. Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane. Proc Natl Acad Sci USA 105:15340–15345.
- Reshetnyak YK, Segala M, Andreev OA, Engelman DM. 2007. A monomeric membrane peptide that lives in three worlds: In solution, attached to, and inserted across lipid bilayers. Biophys J 93:2363–2372.
- Robertson N, Potter C, Harris AL. 2004. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res 64:6160–6165.
- Rofstad EK, Mathiesen B, Kindem K, Galappathi K. 2006. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res 66:6699–6707.
- Samad A, Alam MI, Saxena K. 2009. Dendrimers: A class of polymers in the nanotechnology for the delivery of active pharmaceuticals. Curr Pharm Des 15:2958–2969.
- Schornack PA, Gillies RJ. 2003. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 5:135–145.
- Schroeder A, Kost J, Barenholz Y. 2009. Ultrasound, liposomes, and drug delivery: Principles for using ultrasound to control the release of drugs from liposomes. Chem Phys Lipids 162:1–16.
- Semalty A, Semalty M, Rawat BS, Singh D, Rawat MS. 2009. Pharmacosomes: The lipid-based new drug delivery system. Expert Opin Drug Deliv 6:599–612.
- Sennoune SR, Luo D, Martinez-Zaguilan R. 2004. Plasmalemmal vacuolar-type H+-ATPase in cancer biology. Cell Biochem Biophys 40:185–206.
- Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462:55–70.
- Signore A, Annovazzi A, Chianelli M, Corsetti F, Van de Wiele C, Watherhouse RN. 2001. Peptide radiopharmaceuticals for diagnosis and therapy. Eur J Nucl Med 28:1555–1565.
- Srinivas PR, Kramer BS, Srivastava S. 2001. Trends in biomarker research for cancer detection. Lancet Oncol 2:698–704.
- Sun H, Nikolovska-Coleska Z, Yang CY, Qian D, Lu J, Qiu S, Bai L, Peng Y, Cai Q, Wang S. 2008. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc Chem Res 41:1264–1277.
- Tannock IF, Rotin D. 1989. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 49:4373–4384.
- Tekade RK, Kumar PV, Jain NK. 2009. Dendrimers in oncology: An expanding horizon. Chem Rev.
- Thevenin D, An M, Engelman DM. 2009. pHLIP-mediated translocation of membrane-impermeable molecules into cells. Chem Biol 16:754–762.
- Thoren PE, Persson D, Karlsson M, Norden B. 2000. The antennapedia peptide penetratin translocates across lipid bilayers – the first direct observation. FEBS Lett 482:265–268.
- Tsutsumi S, Neckers L. 2007. Extracellular heat shock protein 90: A role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci 98:1536–1539.
- Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. 2004. X-ray structure of a protein-conducting channel. Nature 427:36–44.
- Vaupel P, Kallinowski F, Okunieff P. 1989. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res 49:6449–6465.
- Vavere AL, Biddlecombe GB, Spees WM, Garbow JR, Wijesinghe D, Andreev OA, Engelman DM, Reshetnyak YK, Lewis JS. 2009. A novel technology for the imaging of acidic prostate tumors by positron emission tomography. Cancer Res 69:4510–4516.
- Vives E, Richard JP, Rispal C, Lebleu B. 2003. TAT peptide internalization: Seeking the mechanism of entry. Curr Protein Pept Sci 4:125–132.
- Wang JL, Zhang ZJ, Choksi S, Shan S, Lu Z, Croce CM, Alnemri ES, Korngold R, Huang Z. 2000. Cell permeable Bcl-2 binding peptides: A chemical approach to apoptosis induction in tumor cells. Cancer Res 60:1498–1502.
- Warburg O, Wind F, Negelein E. 1927. The metabolism of tumors in the body. J Gen Physiol 8:519–530.
- Wehland J, Osborn M, Weber K. 1977. Phalloidin-induced actin polymerization in the cytoplasm of cultured cells interferes with cell locomotion and growth. Proc Natl Acad Sci USA 74:5613–5617.
- Wei LY, Roepe PD. 1994. Low external pH and osmotic shock increase the expression of human MDR protein. Biochemistry 33:7229–7238.
- White SH, von Heijne G. 2004. The machinery of membrane protein assembly. Curr Opin Struct Biol 14:397–404.
- Williams AC, Collard TJ, Paraskeva C. 1999. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: Implications for clonal selection during colorectal carcinogenesis. Oncogene 18:3199–3204.
- Wimley WC, White SH. 2000. Determining the membrane topology of peptides by fluorescence quenching. Biochemistry 39:161–170.
- Xiong ZG, Pignataro G, Li M, Chang SY, Simon RP. 2008. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol 8:25–32.
- Yoo J, Cui Q. 2008. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys J 94:L61–63.
- Yuan J, Glazer PM. 1998. Mutagenesis induced by the tumor microenvironment. Mutat Res 400:439–446.
- Zoonens M, Reshetnyak YK, Engelman DM. 2008. Bilayer interactions of pHLIP, a peptide that can deliver drugs and target tumors. Biophys J 95:225–235.