Abstract
Nanomedicine as a field has emerged from the early success of nanoparticle-based drug delivery systems, in particular for treatment of cancer, and the advances made in nano- and biotechnology over the past decade. A prerequisite for nanoparticle-based drug delivery systems to be effective is that the drug payload is released at the target site. A large number of drug release strategies have been proposed that can be classified into certain areas. The simplest and most successful strategy so far, probably due to relative simplicity, is based on utilizing certain physico-chemical characteristics of drugs to obtain a slow drug leakage from the formulations after accumulation in the cancerous site. However, this strategy is only applicable to a relatively small range of drugs and cannot be applied to biologicals. Many advanced drug release strategies have therefore been investigated. Such strategies include utilization of heat, light and ultrasound sensitive systems and in particular pH sensitive systems where the lower pH in endosomes induces drug release. Highly interesting are enzyme sensitive systems where over-expressed disease-associated enzymes are utilized to trigger drug release. The enzyme-based strategies are particularly interesting as they require no prior knowledge of the tumour localization. The basis of this review is an evaluation of the current status of drug delivery strategies focused on triggered drug release by disease-associated enzymes. We limit ourselves to reviewing the liposome field, but the concepts and conclusions are equally important for polymer-based systems.
Introduction
In the past decade, the drug delivery field has experienced dramatically increasing interest, partly due to the early development of long circulating liposomes and the success of marketed formulations such as Doxil®. The field of nanomedicine has emerged as an extension of this success and as a consequence of the advancements in nanotechnology that are now providing new possibilities for designing and characterizing nanomaterials for medical applications.
In the first part of the 1990s, long circulating PEGylated liposomes containing high concentrations of cholesterol were shown to be successful in the delivery of drugs such as doxorubicin (Papahadjopoulos et al. Citation1991, Uziely et al. Citation1995, Gabizon et al. Citation2003) where enhanced delivery of the drug to tumour tissue was based on the enhanced permeation and retention (EPR) effect (for good reviews see Maeda & Matsumura Citation1989, Seymour Citation1992, Yuan et al. Citation1994, Barenholz Citation2001). However, it became apparent that these relatively stable formulations were limited in use to certain classes of drugs (in particular anthracyclins) since cisplatin delivery was found to be ineffective in a clinical study due to lack of drug release after accumulation at the tumour site (Harrington et al. Citation2001). This finding motivated the search for delivery systems that are capable of releasing small molecules as well as macromolecules such as proteins and DNA/RNA upon arrival at the target site. The development of drug delivery systems for cancer treatment has been focused on a number of different strategies: (i) Targeted delivery, where surface ligands such as antibodies are introduced to enhance liposome accumulation in cancerous tissue (Jaracz et al. Citation2005, Sofou & Sgouros Citation2008, Kaasgaard & Andresen Citation2010), (ii) Triggered release, where the liposomes are sensitive to either external stimuli or changes in the microenvironment in cancerous tissue such as enzymes or pH (Drummond et al. Citation2000, Andresen et al. Citation2005a, Hatakeyama et al. Citation2007b), (iii) Passive drug release from liposomes that accumulate in the diseased tissue as a consequence of the EPR effect (i.e., without targeting ligands or active release mechanisms) (Barenholz Citation2001). We can classify the first two as advanced drug delivery systems whereas the last is based on the classic principles that e.g., Doxil was built upon. Even though numerous very interesting advanced drug delivery approaches have been pioneered over the last decade, it is without argument that the commercially most successful designs fall under class three. The main reason for this is simplicity. However, the intrinsic limitations of passive release systems, particularly in the increasingly important field of DNA/RNA delivery, require the design of advanced drug delivery systems to fulfill the potential of namomedicine. Unfortunately, the challenges are substantial, in part, due to a lack of knowledge of tumour microenvironment but also how the delivery system is affected by the biological environment after injection, e.g., what are the effects of protein adsorption in relation to triggered release? We can argue that many of the advanced strategies have been highly successful in in vitro cell experiments but have to some extent failed to provide significant treatment benefits in vivo, probably due to a lack of knowledge of the biological parameters. When this is said, there are many reports of highly efficient advanced drug delivery systems (Drummond et al. Citation2000, Needham & Dewhirst Citation2001, Andresen et al. Citation2005a, Cheong et al. Citation2006, Hatakeyama et al. Citation2007b, Semple et al. Citation2010) and a few have gone through clinical trials.
Many successful delivery strategies are based on active targeting using small peptides or antibodies, however, active release strategies have to a large extent remained at research level with few exceptions. Even so, active release strategies are critical for the future development of nanomedicine, therefore important challenges facing this field must be overcome. This review addresses a number of different approaches within each strategy and discusses the main challenges that have to be addressed for enhancing future development. The main focus of the review is on active release strategies based on enzymatic sensitivity and the most important advances and challenges in the utilization of tumour-associated enzymes in liposome-based drug delivery are discussed.
Advanced drug delivery systems: Targeting and triggering
All drug delivery systems that are developed for intravenous administration have to fulfil a range of general requirements to be successful. We can broadly classify these requirements into: (i) high stability, i.e., shelf-life and during blood circulation, (ii) high accumulation at disease target site, (iii) efficient drug release at the right target site that matches drug pharmacodynamics (i.e., spatial and kinetic control), and (iv) tolerability. Both shelf-life and blood circulation stability (i.e., low degree of lipid hydrolysis, size-, morphology- and drug retention) are strongly influenced by the physico-chemical characteristics of the encapsulated drug and the encapsulation method. For example, the drug loading stability of small molecule chemotherapeutics is enhanced dramatically by using remote loading to give either amorphous precipitate or crystallized drug inside the liposome carrier (Haran et al. Citation1993, Barenholz Citation2001). The encapsulation method furthermore influences the drug release profile and the choice of drug for a given drug delivery strategy should therefore not only depend on the drug efficacy but very much also on its suitability to the drug carrier and release strategy.
Active targeting
Active targeting to tumours involves the attachment of antibodies, peptides or small high affinity ligands to the surface of liposomes with the aim to increase the degree of liposome accumulation in tumours and/or obtain internalization by the cancerous cells. Thus, the liposomes are in principle designed not to rely on passive accumulation as a consequence of the EPR effect but to actively bind to the tumour and cancerous cells through ligand-receptor interactions. However, active targeting is not the focus of the present review and only general concepts will be discussed. Many good reviews already exist in this area (Jaracz et al. Citation2005, Sofou & Sgouros, Citation2008, Kaasgaard & Andresen Citation2010). Generally, there are two active targeting principles that can be exploited: (i) targeting to receptors on endothelial cells that are over-expressed as part of tumour angiogenesis, (ii) targeting to receptors that are over-expressed on the surface of cancerous cells. It is important to realize that the latter strategy is also relying on an initial passive accumulation by the EPR effect and thereby a long blood circulation half-life of the carrier. Thus, this strategy is mainly advantageous for inducing cellular internalization of the carrier prior to drug release and it is necessary to limit the effect on the long circulating properties of the carrier when attaching surface ligands (Allen et al. Citation1995, Andresen et al. Citation2005a, Yamada et al. Citation2008). This is a challenge as many ligands, such as antibodies, are immunogenic when presented on the surface of liposomes and results in lowered blood circulation half-life.
Although the active targeting strategies may seem promising, there are conceptual obstacles that need to be addressed as improved therapeutic efficacy is not necessarily a direct consequence of increased carrier accumulation at the target site. Currently, there is controversy and general contradiction in the literature regarding whether active targeting to receptors on cancerous cells is actually leading to increased accumulation; indeed the majority of data seems to support that this is not usually the case (Gabizon Citation2001, Gabizon et al. Citation2004, Andresen et al. Citation2005a, Kirpotin et al. Citation2006). Furthermore, reports seem to support the view that active targeting to cancerous cells leads to blockage of accumulation and low tumour penetration (Barenholz, Citation2001) since the targeted liposomes bind with high affinity to the first line of cells that is encountered after extravasation and impede further extravasation and tumour infiltration (Emanuel et al. Citation1996). Thus, it may be necessary to optimize the binding affinity of the targeting ligands to cell surface receptors so that binding is not too strong in order to enable deeper tumour penetration. A large ligand-receptor binding constant furthermore means that a high degree of accumulation can only be achieved if the internalization of liposomes into the cancerous cells occurs at a fast rate leaving room for more liposomes to accumulate in the extracellular space in tumours. However, this does not generally seem to be the case.
Active release strategies
The use of tumour-specific targeting does not by itself lead to improved efficacy of a given drug because drug bioavailability at the tumour site is not only dependent on liposome tumour accumulation, but also on the drug release rate from the liposome carrier. The use of targeting ligands may even promote a destructive fate of the liposomal drug within target cells. The receptor targeting strategies directed against the surface of tumour cells can lead to internalization of the liposomes by endocytosis. The endosomes transport their cargo to lysosomes, which may result in degradation of the carried drug if it does not escape the harsh endosomal/lysosomal environment (Gerasimov et al. Citation1999). Consequently, liposomes have to be designed either to escape the endosomes after cell internalization or to release their cargo outside the cell. It is highly drug dependent which of these strategies that is most viable, as drug degradation rate depends on chemical stability, e.g., anthracyclines are very stable in acid and have a relatively long half-life in endosomes/lysosomes. The chemical and metabolic stability of the drugs is therefore very important and should be considered in relation to active targeting and triggering strategies. Several strategies have been proposed to accomplish site-specific triggered drug release in tumour tissue. Liposomes triggered by acid (Yatvin et al. Citation1980, Connor et al. Citation1984, Ellens et al. Citation1984, Collins et al. Citation1990, Ishida et al. Citation2001, Venugopalan et al. Citation2002, Shin et al. Citation2003), small changes in temperature (Yatvin et al. Citation1978, Weinstein et al. Citation1979, Gaber et al. Citation1995, Gaber et al. Citation1996, Kong et al. Citation2000, Needham et al. Citation2000), light (Lamparski et al. Citation1992, Miller et al. Citation1996, Bisby et al. Citation2000, Yavlovich et al. Citation2009) and ultrasound (Huang Citation2008, Schroeder et al. Citation2009, Lentacker et al. Citation2010), have all been shown to be useful concepts for releasing encapsulated drugs. However, liposomes designed with these specific trigger mechanisms have not yet reached the pharmaceutical market. A more recently proposed principle for site-specific drug release is the enzymatically-triggered approaches which are the focus of this review.
Enzyme-triggered release in cancer treatment
The proposed strategies utilizing over-expressed enzymes in tumours are all based on extracellular drug release and can be based on passive tumour accumulation exploiting the EPR effect and potentially also active targeting (). There is substantial knowledge of tumour biology with respect to enzyme over-expression in the extracellular environment of diseased tissue in comparison to healthy tissue. There may be other enzymes of interest in intracellular compartments that can be utilized for drug release but the general viability of such a strategy has not yet been shown. It may also be that there is a not sufficient difference in intracellular enzyme concentrations in cancerous cells relative to normal cells, but currently this knowledge is not available in a drug delivery context. However, by combining targeting of over-expressed receptors that induce cellular internalization with enzymatically-degradable liposomes, it may be possible to create highly efficient new systems.
Figure 1. Illustration of tumour blood vessel fenestration due to the tumour growth and inflammatory condition in this tissue. The liposomes circulate in the blood and extravasate into the extracellular space through pores between the endothelial cells lining the blood vessel. Here the liposomes can encounter secreted enzymes such as sPLA2 or MMPs that then hydrolyze membrane moieties and induce drug release.
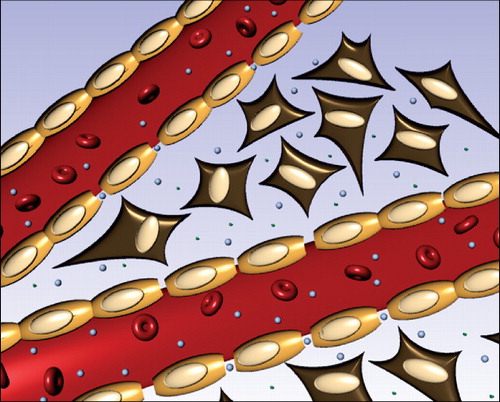
Nonetheless, all successful strategies have been based on secretory enzymes where drug release occurs in the extracellular compartment in the tumour.
Phospholipases
In relation to the development of liposomal drug delivery systems, secretory phospholipase A2 (sPLA2) is so far the only exploited lipase with few exceptions, e.g., the use of lipase secreting bacteria in cancer therapy (Cheong et al. Citation2007). Thus, sPLA2 is a particularly interesting target in drug delivery due to its over-expression in cancerous tissue (Yamashita et al. Citation1993, Abe et al. Citation1997). sPLA2 catalyzes the hydrolysis of phospholipids in the sn-2 position, generating free fatty acids and lysophospholipids. It belongs to the PLA2 superfamily, which comprises a diverse family of lipolytic enzymes that is subdivided into a group of large intracellular lipases: cytosolic 85 kDa Ca2+-sensitive PLA2 (cPLA2), and Ca2+-independent PLA2 (iPLA2); and a group of small secretory 14-19 kDa PLA2 (sPLA2) (Kudo & Murakami Citation2002). To date, ten members (IB, IIA, IIC, IID, IIE, IIF, III, V, X and XIIA) of the sPLA2 family have been indentified in humans (Kudo & Murakami Citation2002), with sPLA2 type IIA (sPLA2-IIA) being the most extensively studied and occurring in the highest concentrations in cancerous tissue. The sPLA2 subgroup shares several characteristics as they all have a low molecular weight ranging from 14–19 kDa, require Ca2+ for enzymatic activation, have a high number of disulfide bonds making them resistant to heat, and are secreted from cells (Berg et al. Citation2001, Laye & Gill Citation2003).
Interestingly, the catalytic activity of sPLA2 is higher towards aggregated phospholipids such as liposomes and micelles than towards lipid monomers, and the activity is highly dependent on membrane charge, lipid composition and the physical state of the lipids. As a consequence, it is possible to design liposomes to be more or less susceptible to sPLA2 hydrolysis by appropriate choice of the phospholipids making up the liposomes. The sPLA2-IIA subtype, is mainly active towards anionic phospholipid membranes and binds several orders of magnitude more tightly to membranes with a net negative charge compared to zwitterionic lipid membranes (Leidy et al. Citation2006). Although a large number of studies have shown that sPLA2 is present and upregulated in cancerous tissue, it is still being debated whether it has a pro- or anti-tumourigenic role in human cancer development (Graff et al. Citation2001, Leung et al. Citation2002). Over-expression of sPLA2-IIA has in particular been identified in a variety of cancer types (Abe et al. Citation1997) including prostate (Jiang et al. Citation2002), colon (Edhemovic et al. Citation2001), breast (Yamashita et al. Citation1993, Yamashita et al. Citation1994) and pancreatic cancer (Kashiwagi et al. Citation1999). It has been suggested that the sPLA2-IIA over-expression in cancerous tissue may be triggered by the carcinoma cells, hepatocytes and/or inflammatory cells (e.g., macrophages) in response to stimulation of inflammatory cytokines (Abe et al. Citation1997). The elevated sPLA2 levels in cancerous tissue suggest that sPLA2 may be a potential target for therapeutic intervention (Laye & Gill Citation2003, Andresen et al. Citation2004a).
The first finding of major importance for the utilization of sPLA2 as a drug release trigger from liposomes was that sPLA2 is more active towards PEGylated liposomes than non-PEGylated liposomes (Jorgensen et al. Citation1999, Jorgensen et al. Citation2002). In the first papers published on this finding, the origin of this increased activity was not clear. However, a study by Andresen et al. (Andresen et al. Citation2002) revealed that it is the negative charge on the PEG lipids, e.g., 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000), that causes the increased activity. Investigations of sPLA2 activity towards liposomes with various lipid compositions (Davidsen et al. Citation2003) resulted in formulations that provide rapid release of drugs such as doxorubicin and cisplatin (Andresen et al. Citation2005a). This release is a consequence of the morphological change of the liposome membrane when lysolipids and fatty acids are formed as a consequence of the enzymatic activity (). sPLA2-degradable liposome formulations of doxorubicin were furthermore shown to enhance the cytotoxicity of the drug in a cell study using sPLA2 secreting Colo 205 colon cancer cells and significantly (Andresen et al. Citation2005a) improved its anticancer efficacy in an in vivo breast cancer xenograft model in mice (MT-3 breast cancer model). It has also been demonstrated that sPLA2 hydrolysis and drug release from liposomes with high cholesterol content, (e.g., Doxil) is essentially zero (Andresen et al. Citation2005a), since the enzyme has no activity towards lipid membranes in the liquid ordered phase. When comparing results from liposome systems that are not degradable by sPLA2 with liposome systems that are, it is clear that the enzyme is responsible for the drug release at least in ex vivo cellular studies (Andresen et al. Citation2005a). Furthermore, in a clinical study (Harrington et al. Citation2001) of cisplatin loaded liposomes with the same lipid composition as Doxil, no treatment benefit was observed after intravenous administration. Although the liposomes were found to accumulate in the tumour, the bioavailability of the encapsulated cisplatin was very low due to the stability of this formulation. The lack of drug release in this study highlights the importance of developing active release strategies and the potential of exploiting tumour-specific enzymes for the induction of drug release. A proof of principle experiment, using tumour associated sPLA2, was provided in a MT-3 xenograft model study in mice using sPLA2 degradable liposomes (DSPC/DSPG/DSPE-PEG2000) with encapsulated cisplatin. A significant reduction in tumour growth was obtained, which suggested an increase in cisplatin bioavailability due to sPLA2-mediated degradation of the liposome carrier (Andresen et al. Citation2005a). However, these findings only provide an indirect proof of sPLA2 activity in vivo as the release could also be unspecific. A cisplatin formulation of sPLA2 degradable liposomes has been investigated in a clinical phase I trial, but the results have not been published to date.
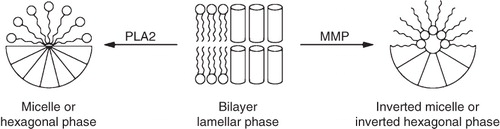
Figure 2. Illustration of how enzymes can induce a morphological change in the liposome membranes giving drug release, or induce transfection with siRNA or DNA plasmid. As an example PLA2 induces a transition to either micelle or a hexagonal phase due to the formation of lysolipids, whereas MMP will reduce headgroup size and induce a transition to an inverted micelle or the inverted hexagonal phase.
Enzyme-activated lipid prodrugs
In addition to the release of drugs encapsulated in the aqueous liposome interior, enzymes can also be used to activate prodrugs that partly constitute the liposome membranes. A number of strategies have been reported focusing on derivatization of drugs to form prodrugs that have high affinity towards the liposome membrane. The prodrug can either be sensitive to chemical hydrolysis or relatively non-specific enzymes such as certain esterases, or be specifically activated by highly specific disease associated enzymes. As an example of the former, Pignatello et al. (Pignatello et al. Citation2003) reported the conjugation of methotrexate (MTX) to lipoamino acids via the carboxyl groups of the glutamate moiety of MTX, and the resulting lipophilic conjugates were incorporated into liposomes. In another study, Kaasgaard et al. (Kaasgaard et al. Citation2009) exploited lipid conjugates of MTX for incorporating MTX prodrugs into the lipid membrane of liposomes that were sensitive to PLA2 hydrolysis. MTX and another weak anticancer agent, docosahexanoic acid (DHA), have also been linked directly to phosphatidylcholine (DHA-MTX-PC) (Zerouga et al. Citation2002). The liposomes incorporating the DHA-MTX-PC construct were reported to be hydrolyzed by snake venom PLA2 giving free MTX and a dose dependent inhibition of murine leukemia cell proliferation. However, no additional studies have been reported on these compounds and their in vivo performance is unknown. Lipid-based prodrug strategies have also been reported for the delivery of mitomycin C (MMC) based on hydrolysable disulfides (Gabizon et al. Citation2006). The prodrug liposome strategy was superior to free MMC against three tumour models with respect to their therapeutic index and antitumour efficiency. The prodrugs were cleverly designed, such that the reduction of the disulfide bridge resulted in an intramolecular elimination to liberate native MMC (Zalipsky et al. Citation2007).
sPLA2 has also been used to release and activate anticancer drugs and prodrugs incorporated into the lipid bilayer. Based on early studies on anticancer ether lipids (AELs) such as edelfosine (Brachwitz & Vollgraf Citation1995, Peters et al. Citation1997, Berkovic Citation1998), a prodrug system was designed that masks the hemolytic properties of AELs (Andresen et al. Citation2004a, Andresen et al. Citation2004b, Jensen et al. Citation2004), and utilizes sPLA2 activity for site-specific activation and liberation of AELs at the tumour site. Several anticancer lipid prodrugs have been synthesized (Andresen et al. Citation2004a, Andresen et al. Citation2004b, Andresen et al. Citation2005b), and were investigated as liposome formulations in Colo205 xenograft models in mice (Jensen et al. Citation2004). However, this first generation of prodrugs was only weakly efficacious at reducing tumour growth in the investigated animal model. This could be due to either too low sPLA2 activity in the tumour extracellular space or low vascularization in the used model, concerns that were not investigated in detail. New generations of prodrugs have been synthesized with structures suitable for attaching more cytotoxic drugs (Linderoth et al. Citation2009, Pedersen et al. Citation2009), and these are now under investigation in cell and animal studies. These sPLA2 strategies offer a new way for designing drug delivery systems, where the drug is situated in the membrane and can be liberated both from the sn-1 or sn-2 position of phospholipids through enzymatic activity. Polyunsaturated fatty acids (Siddiqui et al. Citation2001), retinoids (Altucci & Gronemeyer Citation2001) or prostaglandins (Straus & Glass Citation2001) are some of the more attractive candidates for this strategy.
Proteases
Matrix metalloproteases (MMP) are over-expressed in diseased tissue, e.g., cancerous tissue, and can potentially be exploited as a site-specific trigger of liposome-based drug delivery systems. Different MMP subtypes are over-expressed in different diseases and can be divided into two classes, the transmembrane enzymes of which six subtypes exist, and the extra-cellular subtypes of which 18 exist (Vihinen & Kahari Citation2002, Visse & Nagase Citation2003). In relation to cancer, MMP are thought to play a vital role in invasion, angiogenesis and metastasis (Vihinen et al. Citation2005). The trans-membrane MT1-MMP, the extra-cellular MMP-2 and MMP-9 are in particular over-expressed in many types of human cancers, including brain-, breast-, cervical-, colorectal-, gastric-, lung-, skin-, and ovarian-cancer (Vihinen & Kahari Citation2002, Turpeenniemi-Hujanen Citation2005, Vihinen et al. Citation2005), and have been a target of inhibition strategies for several years. MMP are furthermore a potential target in several other diseases besides cancer, including rheumatoid arthritis, osteoarthritis, periodontitis, and autoimmune blistering disorders of the skin (Vihinen & Kahari Citation2002, Visse & Nagase Citation2003).
The utilization of MMPs for tumour-specific liposomal drug release is not as straightforward as for the PLA2 strategy, since it is necessary to synthesize specialized lipopeptides that are substrates for MMP activation and incorporate these lipopeptides in the liposome membrane. Destabilization of the membrane as a consequence of MMP hydrolysis can be designed in multiple ways. Mallik, Srivastava and co-workers have investigated liposomes containing MMP-9-degradable lipopeptides and shown that MMP-9 can induce liposome destabilization and drug release (Sarkar et al. Citation2005). They developed a peptide mimetic of collagen, the natural substrate of MMP-9 (Briknarova et al. Citation2001, Vihinen & Kahari Citation2002), that forms a triple helical peptide structure through four repeating units of GPO amino acids. In their first paper from 2005 they ‘fish’ the enzymes by utilizing the triple helix as bait and show release of carboxyfluorescein as a result of MMP activity (Sarkar et al. Citation2005) (); However, mechanistic data for the liposome ‘uncorking’ was not provided. Interestingly, the triple helix also prevents non-specific enzymatic activity of trypsin, even though the individual peptide is a substrate of trypsin. In a more recent paper (Elegbede et al. Citation2008) it is shown that drug release upon MMP-9 activation occurs due to lipid mismatch between the lipopeptide and the other lipids that constitute the membrane. The authors have also shown that carboxyfluorescein can be released by incubating the lipopetide containing liposomes in conditioned medium for cancer cells (Banerjee et al. Citation2009) but did not include studies of biological activity, e.g., cytotoxicity studies.
It has long been recognized that selective removal of the PEG coat, which is necessary to obtain long circulating liposomes, specifically in diseased tissue could be a very important step forward in the drug delivery field (Shin et al. Citation2003). Hashida and co-workers (Terada et al. Citation2006) reported the utilization of MMP-2 to unmask PEG coated liposomes. Prior to MMP-2 activation the PEG shield reduced the interaction of galactosylated liposomes with the asialoglycoprotein receptor on the surface of hepatocytes. MMP-2 de-PEGylation provides a way of targeting hepatocellular carcinoma cells specifically due to the secretion of MMP-2 from either cancer or immune cells in cancerous tissue. Using conditioned medium from mouse melanoma cells with high MMP-2 concentration, they showed activation of the galactosylated liposome formulation by MMP-2, which resulted in increased cellular uptake of the liposomes by HepG2 cells.
Harashima and coworkers (Hatakeyama et al. Citation2007b) investigated PEG masked liposomes, using a PEG-peptide-DOPE conjugate, similar in structure to the Hashida lipopeptide (Terada et al. Citation2006), the difference being the lipid anchor. Harashima developed a formulation, stabilized by the PEG-peptide-DOPE conjugates, which was activated by MMP-2 leading to destabilization and liposome fusion/aggregation, as a consequence of the formation of lipids that prefer an inverted hexagonal phase (). They showed that their PEG-peptide-DOPE conjugates could be used to create a promising in vivo transfection system, however, the liposomes were considerably less stable during blood circulation when compared to conventional PEG liposomes and optimization was needed. In a recent study, Harashima and coworkers (Hatakeyama et al. Citation2009) reported an improvement to the system by incorporating pH sensitive GALA lipid conjugates (Nicol et al. Citation1996) and showed that siRNA silencing was higher for the GALA containing liposomes in a HT1080 xenograft model (expressing MMP-2). The combination of the MMP sensitive lipopeptides with pH sensitive lipopeptides offers a system where MMP-2 activates the liposomes in the cancerous tissue for cellular uptake and the pH sensitivity offers increased endosomal escape after cell internalization. No data was provided on how the incorporation of the GALA-lipid conjugate influences the biodistribution of the liposomes.
However, the idea of using peptidases for site-specific triggering of profusiogenic liposomes is not new. Meers and co-workers (Pak et al. Citation1998) investigated the membrane fusion properties of lipopetide containing liposomes as a function of elastase and proteinase K activity and they were able to show that fusion correlates with enzyme hydrolysis. In another paper it was clearly demonstrated that the fluorescent dye calcein (used as a model compound) was internalized by ECV304 cells as a function of elastase activity (Pak et al. Citation1999).
Other enzymatic approaches
A number of other studies of enzymatically-triggered liposomal drug delivery systems have been reported, but the PLA2 and MMP strategies are by far the most thoroughly investigated and successful approaches so far. For sake of completeness a few of the most important other strategies will be discussed briefly. Davis and Szoka (Davis & Szoka Citation1998) reported a novel strategy based on a phosphatase catalyzed triggering of liposomes that promote transfection with plasmid DNA encoding luciferase. The liposomes were composed of cholesterol phosphate derivatives and (DOPE). The phosphatase activation induced a transition from a lamellar phase to the inverted hexagonal phase () as removal of the phosphate reduces the repulsion between the headgroups. This change in morphology could also be induced by addition of calcium, which is known to decrease the electrostatic repulsion between negatively charged headgroups in the interface region. A similar utilization of change in headgroup electrostatic repulsion is also known from a number of pH sensitive drug delivery strategies, e.g., protonation of the oleic acid or CHEMS headgroups (Duzgunes et al. Citation1985, Drummond et al. Citation2000). The same strategy of inducing a lamellar to inverted hexagonal phase transition by enzymatic hydrolysis was described by Pinnaduwage and Huang (Pinnaduwage & Huang Citation1988). They reported a beta-galactosidase-induced destabilization of liposomes composed of phosphatidylethanolamine and ganglioside GM1, which resulted in effective release of calcein in an enzyme-dependent manner due to enzymatic hydrolysis of the GM1 headgroup. In a similar study, Huang showed liposome triggering by trypsin, also giving calcein release, this time from DOPC liposomes containing glycophorin (Hu et al. Citation1986).
Another enzymatically-activated drug delivery strategy was reported by Menger and Johnston (Menger & Johnston Citation1991). By synthesizing a new class of lipids that were sensitive to acetylcholineesterase, they showed that a very abrupt release of a fluorophore occurred after esterase addition. The esterase hydrolysis induced an intra-molecular cyclization that liberated hexadecanol and a detergent like molecule that is likely to induce micellation of the liposomes above a certain concentration in a similar way to that which occurs in the PLA2-based strategy.
Conclusion
One of the most interesting aspects of the many studies that have been carried out on enzymatically-triggered strategies is that the enzymes seem to be capable of penetrating the liposome PEG layer and can hydrolyze the lipids or peptides below this layer. PEG layers are believed to protect the liposome against protein binding during circulation to some extent, but it is clear from publications on the PLA2 and MMP strategies that hydrolysis occurs. The small 14 kDa PLA2 in fact has enhanced affinity for PEGylated liposomes due to the charge that PEG-lipids introduce to the membrane. It is also notable that the larger sized MMP-2 (72 kDa) and MMP-9 (92 kDa) also penetrate the PEG layer and hydrolyze lipopetides located in the interface region of the liposomes (Terada et al. Citation2006, Hatakeyama et al. Citation2007a). This indicates that enzymes in general are able to hydrolyze substrates hidden under the liposome PEG layer, a crucial prerequisite for using enzymes as triggers of liposomal drug delivery systems. One of the drawbacks of designing liposomes for MMP-2 or MMP-9 activation is the necessity to synthesize lipopeptides, which has a much higher cost than natural lipids, particularly when employing both solid- and solution-phase synthesis to achieve the desired conjugates. However, facile and efficient new synthetic approaches have recently been reported giving PEG-peptide-lipid conjugates exclusively by solid-phase synthesis in high yield (Jølck et al. Citation2010).
The utilization of disease-associated enzymes to trigger drug release is probably the most selective and powerful drug release strategy for achieving effective drug delivery to diseased tissue as it offers a possibility for also hitting metastatic and early stage tumours. Many studies have been carried out and the results are promising. However, one should keep in mind that for none of the systems that have been discussed in this review, has there been proof that the in vivo drug release is correlated with enzyme activity. The enzymes that are targeted have been found to be present in the cancerous cells in high concentration but the immunostaining methods used to visualize the enzymes are not conclusive. Current knowledge is based on the putative presence of the enzymes and not their demonstrated activity towards the liposome systems in vivo. One of the biggest questions in this regard is, do the liposomes change lipid composition during circulation? Maintenance of lipid composition is highly important for the enzymatic activity, but it is conceivable that the liposomal membrane composition could change during circulation in these highly designed materials. This is particularly true for the sPLA2 strategy, but is a potential problem that should be considered for all active triggering approaches described in this review. Future studies should focus on elucidating the molecular basis of drug release and not use the treatment efficiency as the only endpoint in animal models.
In summary, using over-expressed enzymes to trigger drug release specifically in tumour tissue has enormous potential and is an area that does not receive the attention it deserves. Understanding the tumour microenvironment in relation to this strategy and how liposomes may change composition during blood circulation are the most important tasks ahead. In particular, researchers should provide new data on the molecular basis for drug release in vivo even though this is a highly challenging task.
Declaration of interest: Financial support was kindly provided by the Danish Strategic Research Council (NABIIT) ref. 2106-07-0033, the Technical University of Denmark (DTU), and NIHGM087016.
References
- Abe T, Sakamoto K, Kamohara H, Hirano Y, Kuwahara N, Ogawa M. 1997. Group II phospholipase A2 is increased in peritoneal and pleural effusions in patients with various types of cancer. Int J Cancer 74:245–250.
- Allen TM, Brandeis E, Hansen CB, Kao GY, Zalipsky S. 1995. A new strategy for attachment of antibodies to sterically stabilized liposomes resulting in efficient targeting to cancer cells. Biochim Biophys Acta 1237:99–108.
- Altucci L, Gronemeyer H. 2001. The promise of retinoids to fight against cancer. Nat Rev Cancer 1:181–193.
- Andresen TL, Davidsen J, Begtrup M, Mouritsen OG, Jorgensen K. 2004a. Enzymatic release of antitumor ether lipids by specific phospholipase A2 activation of liposome-forming prodrugs. J Med Chem 47:1694–1703.
- Andresen TL, Jensen SS, Jorgensen K. 2005a. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog Lipid Res 44:68–97.
- Andresen TL, Jensen SS, Madsen R, Jorgensen K. 2005b. Synthesis and biological activity of anticancer ether lipids that are specifically released by phospholipase A(2) in tumor tissue. J Med Chem 48:7305–7314.
- Andresen TL, Mouritsen OG, Begtrup M, Jorgensen K. 2002. Phospholipase A2 activity: Dependence on liposome surface charge and polymer coverage. Biophys J 82:148A.
- Andresen TL, Skytte DM, Madsen R. 2004b. Synthesis of anti-tumour phosphatidylinositol analogues from glucose by the use of ring-closing olefin metathesis. Org Biomol Chem 2:2951–2957.
- Banerjee J, Hanson AJ, Gadam B, Elegbede AI, Tobwala S, Ganguly B, Wagh AV, Muhonen WW, Law B, Shabb JB, Srivastava DK, Mallik S. 2009. Release of liposomal contents by cell-secreted matrix metalloproteinase-9. Bioconjugate Chem 20:1332–1339.
- Barenholz Y. 2001. Liposome application: problems and prospects. Curr Opin Colloid Interface Sci 6:66–77.
- Berg OG, Gelb MH, Tsai MD, Jain MK. 2001. Interfacial enzymology: the secreted phospholipase A(2)-paradigm. Chem Rev 101:2613–2654.
- Berkovic D. 1998. Cytotoxic etherphospholipid analogues. Gen Pharmacol 31:511–517.
- Bisby RH, Mead C, Morgan CG. 2000. Active uptake of drugs into photosensitive liposomes and rapid release on UV photolysis. Photochem Photobiol 72:57–61.
- Brachwitz H, Vollgraf C. 1995. Analogs of alkyllysophospholipids: chemistry, effects on the molecular level and their consequences for normal and malignant cells. Pharmacol Ther 66:39–82.
- Briknarova K, Gehrmann M, Banyai L, Tordai H, Patthy L, Llinas M. 2001. Gelatin-binding region of human matrix metalloproteinase-2 – Solution structure, dynamics, and function of the COL-23 two-domain construct. J Biol Chem 276:27613–27621.
- Cheong I, Huang X, Bettegowda C, Diaz LA, Kinzler KW, Zhou SB, Vogelstein B. 2006. A bacterial protein enhances the release and efficacy of liposomal cancer drugs. Science 314:1308–1311.
- Cheong I, Huang X, Thornton K, Diaz LA, Zhou SB. 2007. Targeting cancer with bugs and liposomes: ready, aim, fire. Cancer Res 67:9605–9608.
- Collins D, Litzinger DC, Huang L. 1990. Structural and functional comparisons of pH-sensitive liposomes composed of phosphatidylethanolamine and three different diacylsuccinylglycerols. Biochim Biophys Acta 1025:234–242.
- Connor J, Yatvin MB, Huang L. 1984. pH-sensitive liposomes: acid-induced liposome fusion. Proc Natl Acad Sci USA 81:1715–1718.
- Davidsen J, Jorgensen K, Andresen TL, Mouritsen OG. 2003. Secreted phospholipase A(2) as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochim Biophys Acta 1609:95–101.
- Davis SC, Szoka FC. 1998. Cholesterol phosphate derivatives: synthesis and incorporation into a phosphatase and calcium-sensitive triggered release liposome. Bioconjugate Chem 9:783–92.
- Drummond DC, Zignani M, Leroux J. 2000. Current status of pH-sensitive liposomes in drug delivery. Prog Lipid Res 39:409–460.
- Duzgunes N, Straubinger RM, Baldwin PA, Friend DS, Papahadjopoulos D. 1985. Proton-induced fusion of oleic acid-phosphatidylethanolamine liposomes. Biochemistry 24:3091–3098.
- Edhemovic I, Snoj M, Kljun A, Golouh R. 2001. Immunohistochemical localization of group II phospholipase A2 in the tumours and mucosa of the colon and rectum. Eur J Surg Oncol 27:545–548.
- Elegbede AI, Banerjee J, Hanson AJ, Tobwala S, Ganguli B, Wang RY, Lu XN, Srivastava DK, Mallik S. 2008. Mechanistic studies of the triggered release of liposomal contents by matrix metalloproteinase-9. J Am Chem Soc 130:10633–10642.
- Ellens H, Bentz J, Szoka FC. 1984. pH-induced destabilization of phosphatidylethanolamine-containing liposomes: role of bilayer contact. Biochemistry 23:1532–1538.
- Emanuel N, Kedar E, Bolotin EM, Smorodinsky NI, Barenholz Y. 1996. Targeted delivery of doxorubicin via sterically stabilized immunoliposomes: pharmacokinetics and biodistribution in tumor-bearing mice. Pharm Res 13:861–868.
- Gaber MH, Hong K, Huang SK, Papahadjopoulos D. 1995. Thermosensitive sterically stabilized liposomes: formulation and in vitro studies on mechanism of doxorubicin release by bovine serum and human plasma. Pharm Res 12:1407–1416.
- Gaber MH, Wu NZ, Hong K, Huang SK, Dewhirst MW, Papahadjopoulos D. 1996. Thermosensitive liposomes: extravasation and release of contents in tumor microvascular networks. Int J Radiat Oncol Biol Phys 36:1177–1187.
- Gabizon A, Shmeeda H, Barenholz Y. 2003. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet 42:419–436.
- Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. 2004. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev 56:1177–1192.
- Gabizon AA. 2001. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res 7:223–225.
- Gabizon AA, Tzemach D, Horowitz AT, Shmeeda H, Yeh J, Zalipsky S. 2006. Reduced toxicity and superior therapeutic activity of a mitomycin C lipid-based prodrug incorporated in pegylated liposomes. Clin Cancer Res 12:1913–1920.
- Gerasimov OV, Boomer JA, Qualls MM, Thompson DH. 1999. Cytosolic drug delivery using pH- and light-sensitive liposomes. Adv Drug Del Rev 38:317–338.
- Graff JR, Konicek BW, Deddens JA, Chedid M, Hurst BM, Colligan B, Neubauer BL, Carter HW, Carter JH. 2001. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin Cancer Res 7:3857–3861.
- Haran G, Cohen R, Bar LK, Barenholz Y. 1993. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1151:201–215.
- Harrington KJ, Lewanski CR, Northcote AD, Whittaker J, Wellbank H, Vile RG, Peters AM, Stewart JS. 2001. Phase I-II study of pegylated liposomal cisplatin (SPI-077) in patients with inoperable head and neck cancer. Ann Oncol 12:493–496.
- Hatakeyama H, Akita H, Kogure K, Harashima H. 2007a. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Yakugaku Zasshi 127:1549–1556.
- Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, Ueno M, Kobayashi H, Kikuchi H, Harashima H. 2007b. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther 14:68–77.
- Hatakeyama H, Ito E, Akita H, Oishi M, Nagasaki Y, Futaki S, Harashima H. 2009. A pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivo. J Control Release 139:127–132.
- Hu LR, Ho RJY, Huang L. 1986. Trypsin induced destabilization of liposomes composed of dioleoylphosphatidylethanolamine and glyophorin. Biochem Biophys Res Comm 141:973–8.
- Huang SL. 2008. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev 60:1167–1176.
- Ishida T, Kirchmeier MJ, Moase EH, Zalipsky S, Allen TM. 2001. Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim Biophys Acta 1515:144–158.
- Jaracz S, Chen J, Kuznetsova LV, Ojima L. 2005. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem 13:5043–5054.
- Jensen SS, Andresen TL, Davidsen J, Hoyrup P, Shnyder SD, Bibby MC, Gill JH, Jorgensen K. 2004. Secretory phospholipase A(2) as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol Cancer Ther 3:1451–1458.
- Jiang J, Neubauer BL, Graff JR, Chedid M, Thomas JE, Roehm NW, Zhang S, Eckert GJ, Koch MO, Eble JN, Cheng L. 2002. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am J Pathol 160:667–671.
- Jølck RI, Berg RH, Andresen TL. 2010. Solid-phase synthesis of PEGylated lipopeptides using click chemistry. Bioconjugate Chem 21:807–810.
- Jorgensen K, Davidsen J, Mouritsen OG. 2002. Biophysical mechanisms of phospholipase A2 activation and their use in liposome-based drug delivery. FEBS Lett 531:23–27.
- Jorgensen K, Vermehren C, Mouritsen OG. 1999. Enhancement of phospholipase A2 catalyzed degradation of polymer grafted PEG-liposomes: effects of lipopolymer-concentration and chain-length. Pharm Res 16:1491–1493.
- Kaasgaard T, Andresen TL. 2010. Liposomal cancer therapy: exploiting tumor characteristics. Expert Opin Drug Deliv 7:225–243.
- Kaasgaard T, Andresen TL, Jensen SS, Holte RO, Jensen LT, Jorgensen K. 2009. Liposomes containing alkylated methotrexate analogues for phospholipase A(2) mediated tumor targeted drug delivery. Chem Phys Lipids 157:94–103.
- Kashiwagi M, Friess H, Uhl W, Berberat P, Abou-Shady M, Martignoni M, Anghelacopoulos SE, Zimmermann A, Buchler MW. 1999. Group II and IV phospholipase (A2) are produced in human pancreatic cancer cells and influence prognosis. Gut 45:605–612.
- Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong KL, Nielsen UB, Marks JD, Benz CC, Park JW. 2006. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 66:6732–6740.
- Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, Dewhirst MW. 2000. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer Res 60:6950–6957.
- Kudo I, Murakami M. 2002. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat 68–69:3–58.
- Lamparski H, Liman U, Barry JA, Frankel DA, Ramaswami V, Brown MF, O'Brien DF. 1992. Photoinduced destabilization of liposomes. Biochemistry 31:685–694.
- Laye JP, Gill JH. 2003. Phospholipase A2 expression in tumours: a target for therapeutic intervention? Drug Discov Today 8:710–716.
- Leidy C, Linderoth L, Andresen TL, Mouritsen OG, Jorgensen K, Peters GH. 2006. Domain-induced activation of human phospholipase A(2) type IIA: local versus global lipid composition. Biophys J 90:3165–3175.
- Lentacker I, Geers B, Demeester J, De Smedt SC, Sanders NN. 2010. Design and evaluation of doxorubicin-containing microbubbles for ultrasound-triggered doxorubicin delivery: cytotoxicity and mechanisms involved. Mol Ther 18:101–108.
- Leung SY, Chen X, Chu KM, Yuen ST, Mathy J, Ji J, Chan AS, Li R, Law S, Troyanskaya OG, Tu IP, Wong J, So S, Botstein D, Brown PO. 2002. Phospholipase A2 group IIA expression in gastric adenocarcinoma is associated with prolonged survival and less frequent metastasis. Proc Natl Acad Sci USA 99:16203–16208.
- Linderoth L, Peters GH, Madsen R, Andresen TL. 2009. Drug delivery by an enzyme-mediated cyclization of a lipid prodrug with unique bilayer-formation properties. Angew Chemie Int Ed 48:1823–1826.
- Maeda H, Matsumura Y. 1989. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst 6:193–210.
- Menger FM, Johnston DE. 1991. Specific enzyme-induced decapsulation. J Am Chem Soc 113:5467–5468.
- Miller CR, Bennett DE, Chang DY, O'Brien DF. 1996. Effect of liposomal composition on photoactivated liposome fusion. Biochemistry 35:11782–11790.
- Needham D, Anyarambhatla G, Kong G, Dewhirst MW. 2000. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res 60:1197–1201.
- Needham D, Dewhirst MW. 2001. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv Drug Deliv Rev 53:285–305.
- Nicol F, Nir S, Szoka FC. 1996. Effect of cholesterol and charge on pore formation in bilayer vesicles by a pH-sensitive peptide. Biophys J 71:3288–3301.
- Pak CC, Ali S, Janoff AS, Meers P. 1998. Triggerable liposomal fusion by enzyme cleavage of a novel peptide-lipid conjugate. Biochim Biophys Acta 1372:13–27.
- Pak CC, Erukulla RK, Ahl PL, Janoff AS, Meers P. 1999. Elastase activated liposomal delivery to nucleated cells. Biochim Biophys Acta 1419:111–126.
- Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang SK, Lee KD, Woodle MC, Lasic DD, Redemann C. 1991. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci USA 88:11460–11464.
- Pedersen PJ, Christensen MS, Ruysschaert T, Linderoth L, Andresen TL, Melander F, Mouritsen OG, Madsen R, Clausen MH. 2009. Synthesis and biophysical characterization of chlorambucil anticancer ether lipid prodrugs. J Med Chem 52:3408–3415.
- Peters AC, Ahmad I, Janoff AS, Pushkareva MY, Mayhew E. 1997. Growth inhibitory effects of liposome-associated 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphocholine. Lipids 32:1045–1054.
- Pignatello R, Puleo A, Puglisi G, Vicari L, Messina A. 2003. Effect of liposomal delivery on in vitro antitumor activity of lipophilic conjugates of methotrexate with lipoamino acids. Drug Delivery 10:95–100.
- Pinnaduwage P, Huang L. 1988. Beta-galactosidase-induced destabilization of liposome composed of phosphatidylethanolamine and ganglioside-GM1. Biochim Biophys Acta 939:375–382.
- Sarkar NR, Rosendahl T, Krueger AB, Banerjee AL, Benton K, Mallik S, Srivastava DK. 2005. ‘Uncorking’ of liposomes by matrix metalloproteinase-9. Chem Comm 8:999–1001.
- Schroeder A, Kost J, Barenholz Y. 2009. Ultrasound, liposomes, and drug delivery: principles for using ultrasound to control the release of drugs from liposomes. Chem Phys Lipids 162:1–16.
- Semple SC, Akinc A, Chen JX, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen QM, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ. 2010. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 28:172–176.
- Seymour LW. 1992. Passive tumor targeting of soluble macromolecules and drug conjugates. Crit Rev Ther Drug Carrier Syst 9:135–187.
- Shin J, Shum P, Thompson DH. 2003. Acid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG-lipids. J Control Release 91:187–200.
- Siddiqui RA, Jenski LJ, Neff K, Harvey K, Kovacs RJ, Stillwell W. 2001. Docosahexaenoic acid induces apoptosis in Jurkat cells by a protein phosphatase-mediated process. Biochim Biophys Acta 1499:265–275.
- Sofou S, Sgouros G. 2008. Anti body-targeted liposomes in cancer therapy and imaging. Expert Opin Drug Deliv 5:189–204.
- Straus DS, Glass CK. 2001. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev 21:185–210.
- Terada T, Iwai M, Kawakami S, Yamashita F, Hashida M. 2006. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J Control Release 111:333–342.
- Turpeenniemi-Hujanen T. 2005. Gelatinases (MMP-2 and-9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie 87:287–297.
- Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, Yehoshua Z, Libson E, Muggia FM, Gabizon A. 1995. Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol 13:1777–1785.
- Venugopalan P, Jain S, Sankar S, Singh P, Rawat A, Vyas SP. 2002. pH-sensitive liposomes: mechanism of triggered release to drug and gene delivery prospects. Pharmazie 57:659–671.
- Vihinen P, Ala-aho R, Kahari VM. 2005. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targ 5:203–220.
- Vihinen P, Kahari VM. 2002. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer 99:157–166.
- Visse R, Nagase H. 2003. Matrix metalloproteinases and tissue inhibitors of metalloproteinases – Structure, function, and biochemistry. Circulation Res 92:827–839.
- Weinstein JN, Magin RL, Yatvin MB, Zaharko DS. 1979. Liposomes and local hyperthermia: selective delivery of methotrexate to heated tumors. Science 204:188–191.
- Yamada A, Taniguchi Y, Kawano K, Honda T, Hattori Y, Maitani Y. 2008. Design of folate-linked liposomal doxorubicin to its antitumor effect in mice. Clin Cancer Res 14:8161–8168.
- Yamashita S, Ogawa M, Sakamoto K, Abe T, Arakawa H, Yamashita J. 1994. Elevation of serum group II phospholipase A2 levels in patients with advanced cancer. Clin Chim Acta 228:91–99.
- Yamashita S, Yamashita J, Sakamoto K, Inada K, Nakashima Y, Murata K, Saishoji T, Nomura K, Ogawa M. 1993. Increased expression of membrane-associated phospholipase A2 shows malignant potential of human breast cancer cells. Cancer 71:3058–3064.
- Yatvin MB, Kreutz W, Horwitz BA, Shinitzky M. 1980. pH-sensitive liposomes: possible clinical implications. Science 210:1253–1255.
- Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. 1978. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 202:1290–1293.
- Yavlovich A, Singh A, Tarasov S, Capala J, Blumenthal R, Puri A. 2009. Design of liposomes containing photopolymerizable phospholipids for triggered release of contents. J Therm Anal Calorim 98:97–104.
- Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. 1994. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res 54:3352–3356.
- Zalipsky S, Saad M, Kiwan R, Ber E, Yu N, Minko T. 2007. Antitumor activity of new liposomal prodrug of mitomycin C in multidrug resistant solid tumor: Insights of the mechanism of action. J Drug Target 15:518–530.
- Zerouga M, Stillwell W, Jenski LJ. 2002. Synthesis of a novel phosphatidylcholine conjugated to docosahexaenoic acid and methotrexate that inhibits cell proliferation. Anticancer Drugs 13:301–311.