Abstract
Membrane proteins represent up to 30% of the proteins in all organisms, they are involved in many biological processes and are the molecular targets for around 50% of validated drugs. Despite this, membrane proteins represent less than 1% of all high-resolution protein structures due to various challenges associated with applying the main biophysical techniques used for protein structure determination. Recent years have seen an explosion in the number of high-resolution structures of membrane proteins determined by NMR spectroscopy, especially for those with multiple transmembrane-spanning segments. This is a review of the structures of polytopic integral membrane proteins determined by NMR spectroscopy up to the end of the year 2010, which includes both β-barrel and α-helical proteins from a number of different organisms and with a range in types of function. It also considers the challenges associated with performing structural studies by NMR spectroscopy on membrane proteins and how some of these have been overcome, along with its exciting potential for contributing new knowledge about the molecular mechanisms of membrane proteins, their roles in human disease, and for assisting drug design.
Introduction
Membrane proteins are coded by up to 30% of the open reading frames in known genomes (Wallin and von Heijne Citation1998, Liu and Rost Citation2001, Fagerberg et al. Citation2010) and play important roles in many biological processes including transport of ions and molecules, control of transmembrane potential, the generation and transduction of energy, signal recognition and transduction, and catalysis of chemical reactions. Mutations in membrane proteins are linked to many human diseases (von Heijne Citation2007, Watanabe et al. Citation2008, Rosenbaum et al. Citation2009) and they have been estimated to be the molecular targets for around 50% of validated drugs (Drews Citation2000, Hopkins and Groom Citation2002, Overington et al. Citation2006, Lundstrom Citation2006) whilst also remaining a principal target for new drug discovery in the treatment of a wide range of disorders including cancer, heart disease, obesity, nervous system disorders, inflammation and pain, asthma, cystic fibrosis, Parkinson's disease and the resistance to antibiotics (von Heijne Citation2007, Largerström and Schiöth Citation2008, Müller et al. Citation2008, Arinaminpathy et al. Citation2009, Congreve and Marshall Citation2010). Despite all this, the number of high-resolution structures of membrane proteins in the Protein Data Bank (PDB) is less than 1% of the total for all proteins, therefore limiting the information available for understanding the molecular mechanisms of biological processes involving membrane proteins and for drug design.
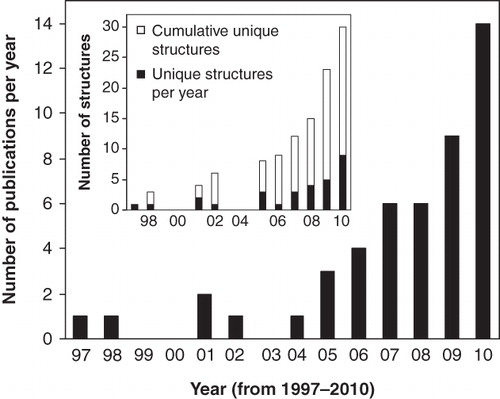
The relatively few structures of membrane proteins, compared with non-membrane proteins, comes from difficulties in their expression and purification and achieving reconstitution conditions that retain their native structure and function whilst also being compatible with the main biophysical techniques used for structure determination: X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. At the end of the year 2010 the number of structures of unique membrane proteins in the PDB was 263 (out of over 60,000 protein structures) with the large majority determined by crystallography. The past few years, however, have seen an explosion in the number of membrane protein structures determined by NMR spectroscopy, especially for those with multiple transmembrane-spanning segments ().
Figure 1. Numbers of published polytopic integral membrane protein structures determined by NMR spectroscopy up to the end of the year 2010.
This is a review of the high-resolution NMR structures of polytopic integral membrane proteins published up to the end of the year 2010, which includes examples of porins, ion channels, integral membrane enzymes, histidine and tyrosine kinase receptors, G protein-coupled receptors (GPCRs) and proteins involved in cell death, mercury detoxification, platelet aggregation and disulphide bond formation using both solution- and solid-state NMR spectroscopy. It also considers the challenges associated with performing structural studies by NMR spectroscopy on membrane proteins and how some of these have been overcome, along with its exciting potential for contributing new knowledge about the molecular mechanisms of membrane proteins, their roles in human disease, and for assisting drug design.
The primary information that was used to compile this review comes from searches of the databases ISI Web of Knowledge, PubMed (MEDLINE) and the Protein Data Bank (PDB) and using online resources that monitor the published structures of membrane proteins, for example, that from the Stephen White laboratory at UC Irvine: http://blanco.biomol.uci.edu/Membrane_Proteins_xtal (White Citation2009).
Challenges for the determination of membrane protein structures by NMR spectroscopy
Prior to applying NMR experiments to determine the high-resolution structure of a membrane protein, a number of challenges have to be overcome. The main ones are summarized below.
Expression
The first challenge when using any biophysical technique for structural studies on membrane proteins is achieving sufficient quantities of protein, which usually has to be purified. NMR is no exception, as it generally requires milligram quantities of protein, so a suitable recombinant expression system or other method of producing the protein of interest is required. As we will see later, expression in Escherichia coli is still the principal system used to produce membrane proteins for structural studies by NMR; chemical synthesis and cell-free expression have also been successfully used. Eukaryotic membrane proteins are particularly challenging to express so heterologous expression systems using yeast (Pichia pastoris and Saccharomyces cerevisiae), baculovirus-infected insect cells and mammalian cells have been developed (Akermoun et al. Citation2005, Midgett and Madden Citation2007, Koth and Payandeh Citation2009, Hays et al. Citation2010, Tapaneeyakorn et al. Citation2011), but these have not yet produced integral membrane proteins whose full structures have been determined by NMR.
Purification and reconstitution
Solution-state NMR studies almost always require the membrane protein of interest to be removed from its native membrane environment and to be in a purified state. A suitable protocol is therefore required to solubilize the protein from the membrane (usually using a detergent) and to isolate it from other proteins (e.g., by affinity chromatography). Some proteins may be produced in a denatured state as inclusion bodies, which is a way of obtaining large quantities of highly pure protein, but an efficient refolding protocol is then required. Following purification, the membrane protein has to be kept solubilized under conditions that mimic its native membrane environment, thus retaining its native structure and function as far as possible, whilst also being compatible with achieving high-resolution NMR spectra. This has mostly been successful by reconstitution of the protein in detergent micelles or in detergent-lipid bicelles. Deuterated detergents are often used to avoid signal overlap with the protein and/or to improve spectral quality, but these can be expensive or difficult to source. It is also important to have alternative biophysical and/or biochemical methods available to confirm that the sample conditions used for NMR structural studies do retain the native structure and function of the protein, but micelle conditions can interfere with functional assays. Strategies for the preparation of membrane protein samples in detergent micelles and in bicelles for solution-state NMR spectroscopy have been described in a number of reviews (Bader et al. Citation2002, Krueger-Koplin et al. Citation2004, Page et al. Citation2006, Tamm and Liang Citation2006, Sanders and Sönnichsen Citation2006, Poget and Girvin Citation2007, Kim et al. Citation2009).
Solid-state NMR is amenable to a wider range of sample types for membrane proteins; they may be used expressed in their native membranes, purified and reconstituted in detergents or lipids, or in a crystalline or lyophilized state; (see work by Lorch et al. Citation2005, Watts Citation2007, De Angelis and Opella Citation2007, Williamson Citation2009 and Tapaneeyakorn et al. Citation2011 for descriptions of sample preparation strategies for solid-state NMR on membrane proteins).
Achieving high-resolution NMR spectra
Achieving high-resolution NMR spectra for a membrane protein is a prerequisite to applying experiments for assignment of its signals and solving its structure. Many of the most interesting and important membrane proteins, such as transporters and GPCRs, have a mass (∼30–50 kDa) which makes them intractable to the standard solution-state NMR methods routinely used for determining the structures of smaller non-membrane proteins. Beyond a size of ∼30 kDa the tumbling rate (or rotation correlation time) of a protein in solution is slowed down such that line-broadening interactions are not removed sufficiently to achieve high-resolution spectra. This is confounded further for membrane proteins solubilized in detergent where the protein-micelle complex has an even longer correlation time and an effective size that can be three- to five-times higher than the protein itself. A membrane protein with a mass of 10 kDa may therefore be in a protein-micelle complex with a mass of 30 kDa or higher. Larger proteins also have a greater number of residues which increases the chance of signal overlap and the number of assignments and distance restraints that have to be measured to solve the structure. Proteins with an α-helical secondary structure tend to have a narrower chemical shift dispersion compared to that of proteins comprised of β-sheets, which contributes further to signal overlap. Transverse relaxation-optimized spectroscopy (TROSY)-based solution-state NMR experiments were developed to enable high-resolution spectra to be obtained for very large systems not previously amenable to NMR (Pervushin et al. Citation1997, Salzmann et al. Citation1998, Citation1999); these have been successfully used to assist the structure determination of membrane proteins, examples of which are described later. The TRACT (TROSY for correlation times) experiment was developed for measuring the effective rotation correlation times and estimating the molecular weights of large molecules in solution (Lee et al. Citation2006). As with any other types of sample, using higher-field NMR magnets and cryoprobes improves sensitivity and, for TROSY-based experiments, spectral resolution too.
Solid-state NMR has no upper size limit to the size of the complex that can be investigated, as reflected in the range of sample types for membrane proteins that can be used.
Isotope labelling
NMR-observable isotopic labels (usually 13C and 15N) have to be incorporated into the protein of interest. The production of uniformly-labelled protein is generally straightforward, if expression in E. coli using a minimal medium can be achieved, by simply using a 15N-labelled nitrogen source (usually [15N]NH4Cl) and/or a 13C-labelled carbon source (usually [U-13C]glucose or [U-13C]glycerol), which are not too expensive. The production of amino acid-specific selectively labelled protein requires the addition of 13C/15N-labelled amino acids or their precursors into the growth medium, which can be expensive, and there can be problems associated with scrambling of the labels. Deuteration of the protein is often combined with TROSY-based NMR experiments to further improve spectral resolution; this is usually achieved by growth of E. coli in D2O instead of H2O and by using deuterium-labelled glucose as carbon source, which is combined with the 13C/15N-labelling; this can lead to a drastic reduction in protein yield and is also expensive. Isotope labelling of eukaryotic membrane proteins expressed in insect or mammalian cells is even more challenging.
Stability
Solution-state NMR experiments on membrane proteins usually have to be performed with the sample at a temperature of at least 20°C and sometimes up to as much as 60°C. A higher temperature makes the system more mobile, which reduces correlation times and reduces the effective size of the complex, therefore improving spectral resolution. The membrane protein has to be stable at these temperatures for days or weeks to enable the NMR experiments for structure determination to be performed. Using the highest temperatures is not viable for most membrane proteins and some systems, e.g., wild-type GPCRS, have only very short lifetimes even at modest temperatures. It is therefore important to have ways to monitor that the structure and activity of the membrane protein in the NMR sample is retained throughout the period of NMR acquisition.
Solid-state NMR experiments on membrane proteins are usually performed with the sample at temperatures of less than 4°C, so thermal damage to the protein is not such an issue. The experiments are also often performed with the protein in native membranes or in near-native conditions where it is likely to be more stable than in the detergents used for solution-state NMR studies.
Structures of polytopic integral membrane proteins determined by NMR spectroscopy
Details for the structures of polytopic integral membrane proteins determined by NMR spectroscopy up to the end of year 2010 are given in and . The table entries cover 48 structural studies on 30 unique proteins, where uniqueness refers to a different protein or the same protein from a different organism and does not include different-sized fragments of the same protein. The proteins are listed with β-barrel systems first and then α-helical systems in an order of increasing complexity in terms of number of unique transmembrane-spanning segments and then the date published. The 30 proteins represent ∼12% of the total number of unique membrane protein structures in the PDB at the end of 2010 and over 60% of these have come in the last three years. The proteins originate from a range of organisms including bacteria, yeast, humans and viral proteins. The following sections in this review will describe the NMR structures of membrane proteins, especially highlighting where technical challenges have been overcome and where novel methods have been used.
Table I. Details for the structures of polytopic integral membrane proteins determined by NMR spectroscopy published up to the end of the year 2010: Protein information, NMR conditions, PDB entry number.
Table II. Details for the structures of polytopic integral membrane proteins determined by NMR spectroscopy published up to the end of the year 2010: NMR structural restraints. The numbering of the entries in this table corresponds with those in .
β-barrel proteins
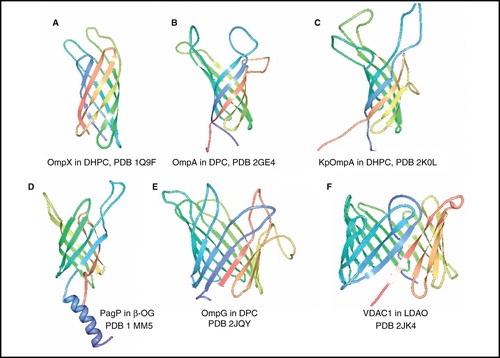
At the end of 2010 there were over 50 structures of unique β-barrel membrane proteins in the PDB, almost all of bacterial origin. NMR structures have been determined for six of these, including five unique bacterial outer membrane proteins and the only β-barrel membrane protein from humans with a known structure ().
Figure 2. NMR structures of β-barrel membrane proteins. Structures of (A) OmpX in DHPC, (B) OmpA from E. coli in DPC, (C) OmpA from Klebsiella pneumoniae in DHPC (D) PagP in β-OG, (E) OmpG in DPC and (F) VDAC1 in LDAO. All pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
Bacterial outer membrane proteins
A number of β-barrel outer membrane proteins from E. coli provided the first high-resolution NMR structures of large integral membrane proteins ( and , entries 1–5, 7, 8) and initiated the application of TROSY-based solution-state NMR experiments to investigate such systems. The first application was in 2001 with the 8-β-strand (148 residues/16.5 kDa) outer membrane protein X (OmpX) reconstituted in 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) micelles (mass of protein-micelle complex ∼ 60 kDa at 30°C) (Fernández et al. Citation2001), which achieved complete sequence-specific assignments and a backbone fold that had good match with the 1.9-Å resolution X-ray crystal structure of OmpX determined in the detergent n-octyltetraoxyethylene (Vogt and Schulz Citation1999). A more complete NMR structure of OmpX was presented in 2004 from a perdeuterated OmpX sample with selectively protonated Val, Leu and Ile(δ1) methyl groups, which allowed measurement of a four-fold increase in the number of nuclear overhauser enhancements (NOEs) between methyl groups and from methyl groups to amide protons (Fernández et al. Citation2004) (). This was the first application of so-called ILV-labelling to assist the structure-determination of a membrane protein, a method which had been developed by Tugarinov and Kay (Citation2003) on the 723-residue (81 kDa) soluble protein malate synthase G.
The structure of 8-β-strand E. coli outer membrane protein A (OmpA) transmembrane domain (177 residues, 19 kDa) in n-dodecylphosphocholine (DPC) micelles (46 kDa complex at 50°C) was also determined in 2001 using TROSY-based experiments (Arora et al. Citation2001); this was shown to closely match the X-ray crystal structure of OmpA determined in the detergent n-octyltetraoxyethylene (Pautsch and Schulz Citation1998). Refinements of the OmpA structure were presented in 2006 by application of site-directed spin labelling (SDSL) and paramagnetic relaxation enhancement (PRE) for long-range distances (Liang et al. Citation2006) and then residual dipolar couplings (RDCs) from samples aligned in polyacrylamide gels; this increased the backbone accuracy to 1.02 Å relative to the crystal structure (Cierpicki et al. Citation2006) (). In 2007 the structure of OmpA transmembrane domain with shortened extracellular loops in DHPC micelles was examined and it was concluded that the removed amino acid residues are not critical for refolding of the protein in vitro (Johansson et al. Citation2007).
The structure of the larger transmembrane domain (210 residues, 23 kDa) of OmpA from Klebsiella pneumoniae (KpOmpA) was determined in DHPC micelles (60–70 kDa complex at 40°C) (Renault et al. Citation2009) (). This work emphasized the importance of preparing samples with selective protonation of the methyl groups at the γ-positions of valine and the δ-positions of leucine and isoleucine residues in a perdeuterated background and the measurement of methyl to methyl and methyl to backbone amide 1H-1H NOEs for obtaining long range distance constraints and significant improvement in the precision of the NMR structure. The structure of KpOmpA closely resembles those of the crystal and NMR structures of E. coli OmpA, except that the β-strands are on average two residues shorter than those in the crystal structure (Renault et al. Citation2009).
Also with 8-β-strands, the structure of E. coli outer membrane enzyme PagP (170 residues, 20 kDa), which transfers a palmitate chain from a phospholipid to lipid A, was determined in both DPC (50–60 kDa complex at 45°C) and n-octyl-β-D-glucoside (β-OG) micelles (Hwang et al. Citation2002) (). Unlike in OmpX and OmpA, the antiparallel β-barrel in PagP is preceded by an amphipathic N-terminal α-helix, which is thought to lie on the periplasmic surface of the membrane. It is worthy to note that this structure of PagP was the first determined for a β-barrel membrane protein before a crystal structure of the protein was available.
The largest bacterial β-barrel membrane protein for which an NMR structure has been determined to date is the 14-β-strand E. coli outer membrane protein G (OmpG) (280 residues, 33 kDa), which facilitates the uptake of large oligosaccharides when the primary facilitator LamB is defective or down-regulated. The NMR structure in DPC micelles (Liang and Tamm Citation2007) () is in good agreement in the β-barrel region with crystal structures of OmpG in the open state (in n-octyltetraoxyethylene at pH 5.5 and in lauryldimethylamine N-oxide (LDAO) at pH 7.5) and closed state (in β-OG at pH 5.6) (Subbarao and van den Berg Citation2006, Yildiz et al. Citation2006), except that the barrel is shorter and more dynamic towards the extracellular side in the NMR structure. The NMR structure was obtained at a pH of 6.3 and is thought to represent a mixture of open and closed conformers that are undergoing exchange on the NMR timescale.
The bacterial β-barrel outer membrane proteins for which NMR structures have been determined (and those from crystal structures) are all monomeric, they all have an even number of antiparallel β-strands, the N- and C-termini are both at the periplasmic side of the membrane, and the extracellular loops are generally longer than the periplasmic loops.
Human voltage-dependent anion channel (VDAC1)
The first and only eukaryotic β-barrel membrane protein for which an NMR structure has been determined is isoform 1 of the human voltage-dependent anion channel (VDAC1) (283 residues, 32 kDa) ( and , entries 9 and 10); this is the most abundant protein in the mitochondrial outer membrane where it mediates the trafficking of small molecules and ions and is involved in mitochondrially induced apoptosis. Two NMR structures of VDAC1 were reported in 2008, both with the protein in LDAO micelles. One structure was obtained by NMR spectroscopy alone (protein-micelle complex 75–90 kDa at 30°C) (Hiller et al. Citation2008) and the other was from a combined NMR/X-ray crystallography study (Bayrhuber et al. Citation2008) (). The structures were in very close agreement with each other and with an X-ray crystal structure of mouse VDAC1 determined in 1,2-dimyristoyl-sn-glycero-3-phosphorylcholine (DMPC)/3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulphonate (CHAPSO) bicelles, also in the same year (Ujwal et al. Citation2008). Unlike all of the structures of bacterial β-barrel membrane proteins that are known (over 50), which have an even number of β-strands and are exclusively antiparallel, VDAC1 has 19 β-strands with the first and last strand parallel and the distribution of longer loops to the extracellular side of the membrane is less distinct, it therefore represents a new class of β-barrel membrane protein fold.
A review by Hiller and Wagner compares the three high-resolution structures of VDAC1 and also considers the role of solution-state NMR for structure-determination of membrane proteins at the time of the VDAC1 structures (Hiller and Wagner Citation2009).
α-helical proteins
At the end of 2010 there were NMR structures for 24 unique α-helical membrane proteins with at least two transmembrane-spanning helices in the PDB. The following sections will describe these in order of increasing number of unique helices and complexity.
Proteins with multiple identical α-helices
The structures of a range of proteins with multiple identical transmembrane-spanning α-helices have been solved and their molecular mechanisms investigated by both solution- and solid-state NMR spectroscopy ( and , entries 11–28).
Non-covalent homodimers
Several years before the advent of TROSY-based experiments, the structure of the non-covalent dimeric transmembrane domain of the human erythrocyte surface protein glycophorin A (40 residues, 9 kDa) in DPC micelles was determined by NMR (MacKenzie et al. Citation1997) (). This landmark study, using just NOEs and J coupling-derived dihedral restraints to solve the structure, demonstrated that van der Waals interactions alone can stabilize specific dimerization of transmembrane helices and showed the potential of NMR for determining the structures of detergent-solubilized α-helical transmembrane-spanning proteins.
Figure 3. NMR structures of α-helical membrane proteins with two identical α-helices (homodimers). Structures of Glycophorin A in DPC, ζζ TM domain (28–60) in SDS, BNip3 TM domain (154–188) in DPC/DPPC, ErbB2 TM domain (641–684) in DMPC/DHPC, ErbB1/ErbB2 TM domain (634–677/641–684) in DMPC/DHPC, EphA1 TM domain (536–573) in DMPC/DHPC, EphA2 TM domain (523–563) in DMPC/DHPC. All pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
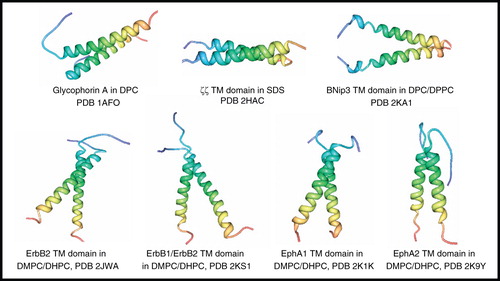
The structures of six further systems with two identical non-covalent α-helices (homodimers) from humans have been solved by solution-state NMR methods ().
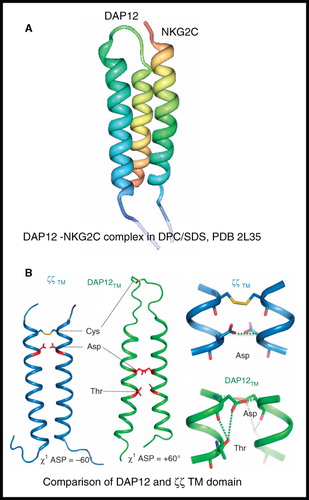
The structure of the ζζ (zetazeta) transmembrane domain (33 residues, 7.5 kDa) in 5:1 DPC/sodium dodecyl sulphate (SDS) mixed micelles (Call et al. Citation2006) demonstrated significant differences in its conformation and packing interactions compared with those for the glycophorin A dimer. Whilst the glycophorin A dimer is a right-handed coiled coil with a crossing angle of −40° and interchain contacts come from 13 out of 23 transmembrane residues, the ζζ dimer forms a left-handed coiled coil with a less severe crossing angle of +23° and interchain contacts come from 20 out of 23 transmembrane residues. The work on the ζζ dimer emphasized the importance in accurate determination of side-chain conformation for analysis of helix packing and demonstrated that three polar residues are crucial for ζζ dimerization and its assembly with the T cell receptor.
BNip3 is an apoptotic Bcl-2 protein involved in hypoxic cell death; transition from respiratory to glycolytic metabolism leads to insertion of homodimeric BNip3 transmembrane domain into the outer mitochondrial membrane triggering a cell-death cascade. The structure of BNip3 transmembrane domain (45 residues, 9.9 kDa) was solved by NMR in DMPC/dihexanoylphosphatidylcholine (DHPC) bicelles (44 kDa complex at 40°C), which is the first for a Bcl-2 family transmembrane domain, and shows a right-handed parallel symmetric dimer (Bocharov et al. Citation2007). The structure suggests that the BNip3 dimer is able to conduct ions, which is based on a hydrogen bond-rich His-Ser node in the middle of the membrane, accessibility of the node to water, and a hydrophilic track along the entire length of the membrane-spanning region; this led to the proposal for a unique BNip3-mediated cell death pathway (Bocharov et al. Citation2007). A further NMR study of BNip3 transmembrane domain in DPC micelles with added phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), which stabilized the peptide conformation and improved the quality of the spectra, showed no evidence for a pore or channel (Sulistijo and MacKenzie Citation2009).
The structure of the homodimeric transmembrane domain of the tyrosine kinase growth factor receptor ErbB2 (44 residues, 9.5 kDa) was determined in DMPC/DHPC bicelles (44 kDa complex at 40°C) showing a right-handed symmetrical dimer with α-helices crossing at an angle of −42° in a conformation that is thought to represent the active state of the tyrosine kinase (Bocharov et al. Citation2008a). This was followed by the structure of a heterodimer formed from the transmembrane domains of ErbB1 and ErbB2 (44 residues, 9.5 kDa) in DMPC/DHPC bicelles (∼44 kDa complex at 40°C) in which the capability for multiple polar interactions and hydrogen bonding between transmembrane segments is consistent with the highest affinity for any ErbB dimer (Mineev et al. Citation2010). Structures of the homodimeric transmembrane domains of the tyrosine kinase receptors EphA1 (38 residues, 7.8 kDa) and EphA2 (41 residues, 8.6 kDa) in DMPC/DHPC bicelles were similarly determined (Bocharov et al. Citation2008b, Citation2010). Whilst EphA1 is a right-handed helix bundle, EphA2 is left-handed and therefore demonstrates helix-packing diversity within this family of tyrosine kinase receptors.
M2 proton channel of influenza A virus
A series of elegant solution- and solid-state NMR studies have been performed on the M2 proton channel of influenza A virus to determine its structure and to investigate its mechanism of action and the binding of inhibitors ( and , entries 19–26). The M2 protein (97 amino acid residues) has a single transmembrane α-helix that functions as a homotetrameric proton channel in the viral lipid envelope and is involved in the regulation of pH at various stages of viral replication. pH sensing is provided by His37 and gating is performed by Trp41 (Pinto et al. Citation1997, Tang et al. Citation2002). The proton conductance of M2 is blocked by low micromolar concentrations of the antiviral drugs amantadine and rimantadine, and amantadine has been shown to bind in a 1:1 ratio with the tetramer (Wang et al. Citation1993, Czabotar et al. Citation2004).
A model for the backbone structure of the amantadine-blocked tetrameric transmembrane domain (residues 26–43) of the M2 proton channel was constructed from solid-state NMR experiments on the monomer in DMPC/1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) bilayers and in the presence of amantadine (Hu et al. Citation2007) (). The measurements were performed on uniformly- and selectively-labelled monomer aligned on glass plates and using the NMR experiment polarization inversion spin exchange at the magic angle (PISEMA) (Wu et al. Citation1994), which provides information on the rotation angle and tilt angle of α-helices. The backbone structure of the monomer showed a kink at Gly34 that breaks the α-helical hydrogen bonding in this region. The tetrameric model is a left-handed helical bundle with C4 symmetry and with an average pore diameter of 10 Å; there is a widening of the pore towards the C-terminus of the channel. A proposed radially symmetric position for bound amantadine was shown, but there was no experimental data for the positioning of amantadine.
Figure 4. NMR structures of M2 proton channel from influenza A virus. Structures of (A) residues 26–43 + amantadine in DMPC/DMPG by solid-state NMR, (B) residues 18–60 + rimantadine in DHPC by solution-state NMR showing four molecules of bound rimantadine, (C) residues 18–60, S31N + rimantadine in DHPC by solution-state NMR, (D) residues 18–60, V27A by solution-state NMR, (E) residues 22–46 + amantadine in DLPC by solid-state NMR showing a single molecule of amantadine in its high-affinity site and (F) residues 22–62 in DOPC/DOPE by solid-state NMR. Pictures A–D and F were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). Picture E is adapted by permission from Macmillan Publishers Ltd: Nature (Cady et al. Citation2010; copyright 2010). This Figure is reproduced in colour in Molecular Membrane Biology online.
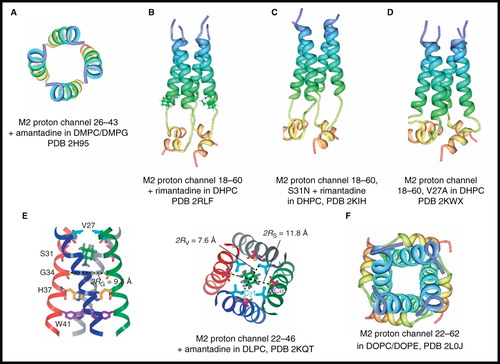
Solution-state NMR was then used to determine the structure of tetrameric M2 (residues 18–60, 20.8 kDa) with bound rimantadine in DHPC micelles (Schnell and Chou Citation2008) (). The structure in the closed state was consistent with a homotetramer in which each subunit has an unstructured N-terminus (residues 18–23), a channel-forming transmembrane helix (25–46), a short flexible loop (47–50) and a C-terminal amphipathic helix (51–59). The transmembrane helices form a left-handed bundle with a narrow channel in which a gate is formed through intermolecular interactions between Trp41 and Asp44 in the adjacent helix. The NMR data led to the proposal for an unexpected external drug-binding site where rimantadine binds at four equivalent sites near the gate on the lipid-facing side of the channel to a polar patch formed by Asp44, Trp41 and Arg45, and that this stabilizes the closed conformation (). An allosteric inhibition mechanism was then described that would account for the effects of drug-resistant mutations. At the same time as this solution-state NMR structure, an X-ray crystal structure was published of the tetrameric transmembrane domain of the M2 proton channel (residues 22–46) in the detergent octyl-β-D-glucopyranoside (Stouffer et al. Citation2008). In contrast with the NMR structure, which showed no binding for an inhibitor molecule inside the pore, the crystal structure showed electron density for the binding of a single molecule of amantadine in a position that physically occludes the pore and is surrounded by residues that are mutated in clinical isolates of amantadine-resistant viruses. It was suggested that the NMR structure ‘represents a biologically relevant closed form of the channel that is unable to bind amantadine in its central cavity’.
Structures of the M2 proton channel had therefore proposed two different sites for the binding of adamantane-based drugs. A solution-state NMR structure of the S31N drug-resistant mutant in DHPC micelles and in the presence of rimantadine () showed that the mutation had little effect on the structure of the channel pore but caused dramatic reduction in drug-binding to the allosteric site with no protein-drug NOEs detected (Pielak et al. Citation2009). The NMR structure along with liposomal proton flux assays were consistent with the proposal that rimantadine inhibition of the M2 proton channel comes from binding at the lipid-facing pocket and were not consistent with the alternative binding site inside the pore. It also suggested that the lipid-facing drug binding site is only present when the helices are tightly packed in the closed conformation and that drug-resistant mutants impair drug binding by destabilising helix-helix assembly.
A solution-state NMR structure of the V27A drug-resistant mutant of the M2 proton channel (residues 18-60) determined in DHPC micelles (Pielak and Chou Citation2010) () was similar to those for the wild-type and S31N mutant, except that it shows a wider channel opening at the N-terminal end and may explain the faster proton conduction for this mutant. Mechanisms for drug-resistance by the V27A mutant via both the allosteric and pore-blocking models were proposed.
A number of solid-state NMR studies were then performed to investigate further the structure of the M2 proton channel and the binding of inhibitors. Magic-angle spinning NMR experiments on M2 (residues 22–46) in 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) lipid bilayers produced a chemical shift-constrained backbone structure for the monomer in the absence and presence of amantadine; this was then used to construct a backbone structure for the amantadine-bound tetramer (Cady et al. Citation2009). The structure in DLPC lipid bilayers was compared with the drug-bound structures determined from 15N static solid-state NMR experiments in DMPC/DMPG bilayers, the solution-state NMR structure in DHPC micelles, and the crystal structure. This highlighted a number of differences including much shorter interhelical Trp41 distances in the static (9.0 Å) and solution NMR (5.9 Å) structures, compared with that measured in DLPC bilayers (16.7 Å). Chemical shift perturbations in the DLPC-embedded channel showed maximum drug-induced changes at residues Ser31 and Val28 and were in favour of an amantadine-binding site inside the N-terminal half of the pore, a similar position to that suggested by the crystal structure. Further magic-angle spinning NMR experiments on M2(22–46) in DMPC bilayers with deuterated amantadine and using the experiment rotational echo double resonance (REDOR) (Gullion and Schaefer Citation1989a, Citation1989b) showed two binding sites for amantadine (Cady et al. Citation2010). The first was a high-affinity site, occupied by a single amantadine, inside the N-terminal of the pore and surrounded by residues mutated in amantadine-resistant viruses; REDOR was used to determine the sub-angstrom resolution structure of the high-affinity site (). The second was a low-affinity site on the C-terminal surface, but only when the drug reaches high concentrations in the bilayer. 2H NMR spectra showed different orientations for amantadine at the two sites, one of the possible orientations at the low affinity site exactly matched that for the rimantadine molecules observed in the solution NMR structure. It was suggested that no drug molecule was observed in the solution NMR structure as it was not isotopically-labelled or it may have been absent from the pore altogether under the micelle conditions. The proton conduction mechanism of the M2 channel was investigated by using solid-state NMR to determine the structure and dynamics of His37 in the TM domain of tetrameric M2 reconstituted into a cholesterol-rich virus-envelope-mimetic lipid membrane (Hu et al. Citation2010). In the high pH closed state, the four histidines form an edge-face π-stacked structure which prevents the formation of a hydrogen-bonded water chain to conduct protons. In the low pH conducting state, the imidazoliums hydrogen-bond extensively with water and undergo microsecond ring reorientations that shuttle protons into the virion. A proton gating and conduction mechanism was proposed for M2 in which ring-flip-assisted imidazole deprotonation is the rate-limiting step. Further solid-state NMR experiments on the M2 conductance domain (residues 22–62) reconstituted in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) lipid bilayers () proposed that the tetrameric His37-Trp41 cluster guides protons through the channel by forming and breaking hydrogen-bonds between adjacent pairs of histidines and through specific interactions of the histidines with the tryptophan gate (Sharma et al. Citation2010). This work also suggests that the hydrophobic pocket at the C-terminus with Asp44 at the bottom is filled by the side chains of Ile51 and Phe54 preventing accessibility to Asp44 and therefore that the external rimantadine binding sites suggested by the solution-state NMR structure are an artefact of the detergent environment.
The work on the M2 proton channel has highlighted the benefits of combining data from solution-state NMR, solid-state NMR and crystallography for determining structure and investigating the molecular mechanisms and the binding of ligands with membrane proteins. In particular this has emphasized the important contribution of solid-state NMR which can perform measurements on membrane proteins in a lipid environment that more closely resembles its native membrane conditions and can determine the high resolution structures and orientations of small-molecule ligands and their binding sites.
A recent review by Wang et al. gives a comprehensive description of the structural and dynamic mechanisms for the function and inhibition of the M2 proton channel coming from studies using a range of biophysical and biochemical techniques (Wang et al. Citation2011).
M2 proton channel of influenza B virus (BM2)
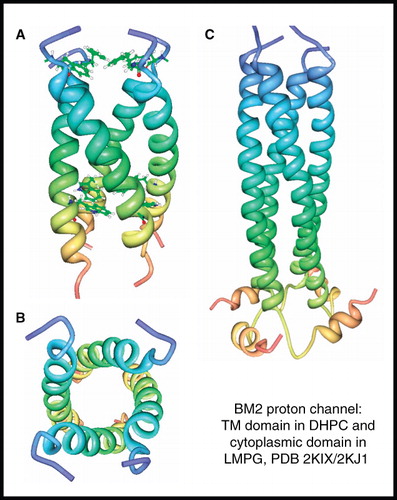
The M2 proton channel of influenza B virus (also known as BM2) is a 109-residue protein that functions as a homotetramer (15.3 kDa) in the viral membrane and is essential for virus replication. BM2 has a different amino acid sequence and channel properties compared with the M2 proton channel of influenza A virus, including an insensitivity to the inhibitors amantadine and rimantadine. BM2 has a membrane-embedded channel domain and a C-terminal cytoplasmic domain, for which NMR structures have been separately determined (Wang et al. Citation2009). The structure of the closed-state (pH 7.5) of the membrane domain (residues 1–33) was determined in DHPC micelles to reveal a coiled-coil tetramer that forms a hydrophilic channel that is occluded by Phe5 and Trp23 at the N- and C-terminal ends, respectively (, ). The structure of the C-terminal extramembrane domain in LMPG micelles showed that residues 45–85 form a helix that oligomerizes into a left-handed coiled-coil tetramer which is connected by a hairpin-like structure, formed by residues 86–92, to a short amphipathic helix (residues 93–103) (). This study also identified polar residues that mediate proton conduction by BM2 and demonstrated that proton conduction by BM2 is about two-fold higher than that of the M2 proton channel from influenza A virus.
Figure 5. NMR structure of BM2 proton channel from influenza B virus. Structure of BM2 transmembrane domain (residues 1–33) in DHPC; (A) view from the membrane plane showing the occluding residues Phe5 and Trp23 and (B) view from the cytosol. Structure of BM2 cytoplasmic domain (residues 26–109) in LMPG (C). These pictures were produced using the PDB files and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
Phospholamban homopentamer
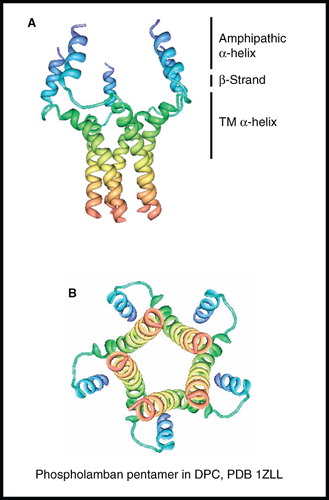
Human phospholamban (52 residues) is expressed in the sarcoplasmic reticulum membrane of muscle cells where it functions as a homopentamer (31 kDa) to control the cellular levels of calcium ions, especially for the regulation of heart muscle contraction and relaxation, involving a mechanism that depends on its phosphorylation. The NMR structure of unphosphorylated phospholamban pentamer was determined in DPC micelles to show a unique ‘bellflower-like’ assembly with five-fold rotational symmetry (Oxenoid and Chou Citation2005) (). Each subunit is made up from a short, positively charged amphipathic α-helix (residues 2–15), an extended linker (residues 16–22) in which residues 18–20 are characteristic of β-strand and a hydrophobic transmembrane α-helix (residues 23–52), which are held together by Leu/Ile zipper interactions in residues 37–51 of the transmembrane part. Adjacent helices cross at an angle of 19° with a left-handed twist. The channel-like structure of phospholamban led to discussion about its potential role as an ion channel and models to propose how its pentameric form could interact with and inhibit the sarco(endo)plasmic reticulum ATPase (SERCA), which pumps calcium ions from the cytoplasm into the sarcoplasmic reticulum. A number of studies on monomeric phospholamban using both solution- and solid-state NMR have also been performed (see Traaseth et al. Citation2009 and references therein).
Figure 6. NMR structure of phospholamban homopentamer. Structure of phospholamban homopentamer in DPC; (A) view from the membrane plane and (B) view from the cytosol. These pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
Proteins with two unique α-helices
A key development was moving from membrane-spanning systems that have multiple identical α-helices, which ‘simplifies’ the spectra, to solving the structures of polytopic membrane proteins with unique α-helices by NMR spectroscopy. The structures of a number of systems with two unique α-helices have now been determined using both solution- and solid-state NMR spectroscopy ( and , entries 29–40).
Subunit c of F1F0 ATP synthase
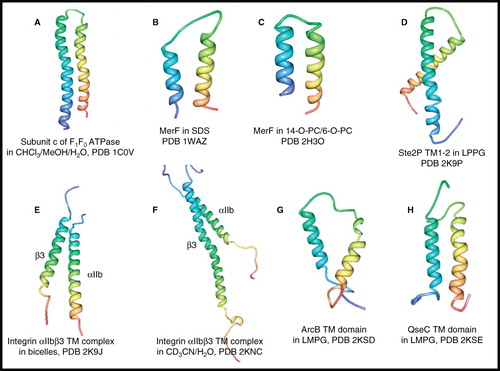
As early as 1998, the structure of the H+-transporting subunit c of F1F0 ATP synthase (79 residues, 8.3 kDa) in a 4:4:1 mixture of chloroform/methanol/water was determined using triple resonance solution-state NMR experiments, including the measurement of NOEs (Girvin et al. Citation1998) (). The structure was described as a hairpin of two antiparallel helical segments with a short connecting loop, as predicted from sequence analysis. Although the solvent mixture used to dissolve the protein leads to high-resolution NMR spectra, caution has to be exercised about its suitability as a membrane mimetic and therefore how the NMR structure of the protein reflects that in its native membrane environment.
Figure 7. NMR structures of α-helical membrane proteins with two unique α-helices. Structures of (A) subunit c of F1F0 ATP synthase in CHCl3/MeOH/H2O, (B) MerF in SDS by solution-state NMR, (C) MerF in 14-O-PC/6-O-PC by solid-state NMR, (D) Ste2P (31–110) TM1-2 of yeast α-factor receptor in LPPG, (E) integrin αIIbβ3 complex in bicelles, (F) integrin αIIbβ3 complex in CD3CN/H2O, (G) ArcB TM domain (1–115) in LMPG, (H) QseC TM domain (1–185) in LMPG. All pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
MerF of the bacterial mercury detoxification system
The first protein with two unique transmembrane α-helices to have its three-dimensional structure determined in a detergent was a truncated form of MerF (residues 13–72, 6.4 kDa) of the mercury detoxification system from the mercury resistant bacterium Morganella morganii in SDS micelles (Howell et al. Citation2005) (). Solution-state NMR measurement of 1H-15N RDCs on samples aligned in stressed polyacrylamide gels and at 60°C allowed backbone structure determination of the helix-loop-helix core domain of the system where residues 27–39 and 50–66 formed the helical segments. The structure also provides a model for full-length wild-type MerF that places two pairs of vicinal cysteine residues (21–22 and 71–72), involved in mercury detoxification, to the cytoplasmic side of the membrane, which is different to previous models for the system. A backbone structure for truncated MerF aligned in phospholipid bicelles was then determined by solid-state NMR spectroscopy (De Angelis et al. Citation2006) (). PISEMA and other double- and triple-resonance experiments were performed on the uniformly- and selectively-labelled protein reconstituted and magnetically-aligned in bicelles formed from 1,2-O-dihexyl-sn-glycero-3-phosphocholine (6-O-PC) and 1,2-O-ditetradecyl-sn-glycero-3-phosphocholine (14-O-PC), which were shown to produce higher resolution spectra compared with the equivalent samples mechanically-aligned on glass plates. The structure was similar to that determined in SDS micelles, except that it had slightly longer helical segments (residues 27–41 and 52–68) and it differed in the N-terminal region (residues 13–24), which is mobile and unstructured in micelles.
The work on MerF was the first to demonstrate the feasibility for determining the structures of polytopic transmembrane systems with non-identical α-helices in both detergent micelles and in phospholipid bicelles by NMR spectroscopy.
Transmembrane helices 1–2 of yeast α-factor GPCR Ste2P
A structure of an 80-residue fragment (31–110, 9.7 kDa) of Ste2P, a GPCR for α-factor receptor of Saccharomyces cerevisiae, was determined in lipopeptidophosphoglycan (LPPG) micelles at 53°C by solution-state NMR (Neumoin et al. Citation2009) (). The fragment composes 19 residues from the N-terminal domain, the first transmembrane helix, the first cytoplasmic loop, the second transmembrane helix and seven residues from the first extracellular loop. This was the first high-resolution NMR structure of a double transmembrane domain fragment of a GPCR in lipid. The structure not only defined the structure of the expected helical hairpin for transmembrane helices 1–2, but revealed an amphipathic helix in the N-terminal tail, which is likely to be surface-associated and may have important implications for GPCR function. This work also further emphasized the benefits of using deuterated detergent/lipid for achieving the highest resolution spectra and the application of ILV methyl-specific labelling for complete structure determination.
Human integrin αIIbβ3 transmembrane complex
The first structure of a heterodimeric transmembrane receptor complex was determined for human integrin αIIbβ3, which is the primary adhesion factor of blood platelets and mediates platelet aggregation by binding to fibrinogen. A complex of the transmembrane domains of αIIb (residues 958–998) and β3 (residues 685–727) (total 84 residues, 9.5 kDa) was formed in phospholipid bicelles and its structure determined by solution-state NMR (Lau et al. Citation2009) (). The structure revealed two dominant integrin transmembrane association motifs, an outer membrane motif characterized by packing interactions centred around three glycine residues, and an inner membrane motif based on unique hydrophobic and electrostatic bridges; these have helped to explain integrin transmembrane signalling. In the same year, a second structure of the integrin αIIbβ3 transmembrane complex formed from αIIb (residues 960–1008) and β3 (residues 689–762) (total 123 residues, 14.8 kDa) in 1:1 acetonitrile/water was determined by solution-state NMR (Yang et al. Citation2009) (). In the transmembrane region the structure revealed a right-handed coiled-coil with the two helices intertwined throughout the membrane and which generally agreed with the structure determined in bicelles. The helices also extended into the cytoplasm and therefore differed significantly in the cytoplasmic tail region compared with the structure determined in bicelles. These differences were attributed to the capture of different conformational states in the acetonitrile/water vs. bicelle environments rather than a result of different construct lengths that were used. The second structure, with the longer cytoplasmic tail present, led to a proposed model for integrin inside-out transmembrane signalling.
Transmembrane domains of histidine kinase receptors ArcB and QseC
All of the systems described so far in this review were produced for NMR experiments by expression in E. coli or by chemical synthesis. A novel approach that combines cell-free protein synthesis, combinatorial dual-isotope labelling and fast acquisition of long-distance information using paramagnetic probes has recently been developed to determine the backbone structures of the transmembrane domains of three different classes of histidine kinase receptors from E. coli (Maslennikov et al. Citation2010). Two of these transmembrane domains, ArcB (115 residues, 13.3 kDa) and QseC (185 residues, 21.3 kDa), are from systems with two unique α-helical segments. These are the first structures (by NMR or crystallography) of the transmembrane domain of any histidine kinase receptors. It was demonstrated on the transmembrane domain of the aerobic respiratory control sensor ArcB that membrane proteins produced by precipitating cell-free expression are comparable to those expressed in E. coli. Verification of fold was achieved by firstly obtaining 13C-13C dipolar-assisted rotational resonance (DARR) (Takegoshi et al. Citation2001) solid-state NMR spectra of the precipitated protein direct from the cell-free synthesis and following its solubilization in the detergent lyso-myristoylphosphatidylglycerol (LMPG) and lyophilization, and then by comparing 1H-15N TROSY spectra for the LMPG-solubilized protein produced by expression using the cell-free system and in E. coli, which proved to be nearly identical. The structures of the ArcB and QseC transmembrane domains (, ) both showed two antiparallel α-helices in which the connecting segment was not well-defined. The quorum sensor QseC has a large periplasmic signalling domain between the two helices, but this was not included in the structure calculations. This work also determined the structure of the transmembrane domain of the K+ sensor KdpD which has four α-helices (see later). A common feature of the three transmembrane domain structures was a loose packing of the α-helical segments and this was attributed to their biological function where inherent flexibility may be essential for signal transmission.
Potassium channel KcsA
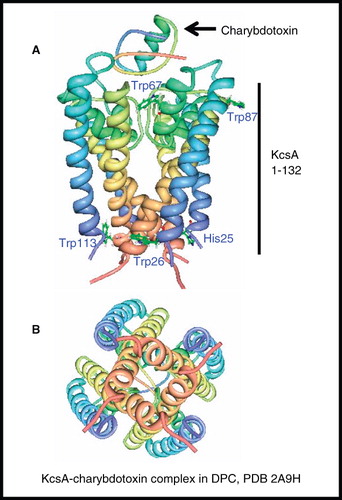
A number of structures have been determined for the homotetrameric (4 × 2 α-helices) potassium channel KcsA from Streptomyces lividans by solution-state NMR. The first was for KcsA (residues 1–132 + 23 N-terminal residues) in complex with the high-affinity peptide antagonist charybdotoxin (37 residues) (total 71 kDa) in DPC micelles using a strategy that relied solely on side chain methyl and aromatic resonances without backbone amide resonance assignment (Yu et al. Citation2005) (, ). This was the first application of ILV methyl-specific labelling in the structure-determination of an α-helical membrane protein. The construct that was used also contained six mutations to mimic critical residues of the eukaryotic Shaker channel and the central cavity of the eukaryotic HERG channel. The NMR structure of the uncomplexed tetramer was entirely consistent with the X-ray crystal structure for the closed conformation of KcsA (residues 23–119) in LDAO, which has C4 symmetry (Doyle et al. Citation1998). The addition of charybdotoxin resulted in chemical shift perturbations in the spectra of KcsA and the detection of intermolecular NOEs to place the binding of the toxin at the extracellular pore of the channel; saturation of the chemical shift perturbations were consistent with binding of one toxin molecule per KcsA tetramer.
Figure 8. NMR structure of potassium channel KcsA. Structure of residues 1–132 in complex with charybdotoxin in DPC; (A) view from the membrane plane also showing residues involved in channel gating, including the pH sensor His25 and (B) view from the cytosol. These pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
The structure of a thermostable N-terminal deletion mutant of KcsA (residues 16–160, 68 kDa) was then determined in SDS micelles (∼ 120 kDa complex at 50°C) (Chill et al. Citation2006), for which the backbone secondary structure was in excellent agreement with the X-ray crystal structure. The C-terminal cytoplasmic domain, which was absent in the crystal structure and earlier NMR structure, was shown to contain a 14-residue α-helix that could participate in tetramerization by forming an intersubunit four-helix bundle. The addition of K+ ions resulted in chemical shift changes in the selectivity filter, and pH-induced changes in chemical shift were observed along the outer transmembrane helix and the cytoplasmic membrane interface.
The effects of pH on the gating and conformation of KcsA was investigated further by NMR experiments on a C-terminal deletion of KcsA (residues 1–124) in n-dodecyl-β-D-maltoside (DDM) micelles (106–132 kDa complex at 50°C) (Takeuchi et al. Citation2007). Chemical shift changes in 1H-15N TROSY spectra going between pH 3.9 (open state) and 5.2 (closed state) represented a conformational arrangement associated with the gating on the intracellular side of the protein; substitution of His25 by Ala abolished this arrangement suggesting that His25 serves as a pH sensor for the channel. The position of His25 is shown on .
The structure of full-length KcsA (residues 1–160) in DPC micelles (∼130 kDa complex at 50°C) in its open, closed and intermediate states was then investigated by NMR (Baker et al. Citation2007). At pH 7 (closed-state) the structure was in agreement with previous crystal and NMR structures, except that the C-terminal helix (residues 131–156) was longer than the one seen for the NMR structure in SDS micelles. This was attributed to the harsher detergent conditions which may partially unfold the cytoplasmic helix. In the open-state (pH 4) the C-terminal helix was more prominent and there were subtle structural changes in the C-terminal half of transmembrane helix 2. The effects of pH titrations on chemical shift values were consistent with conformational changes occurring both at the C-terminus of helix 2 and in the filter when moving between the closed and open states. The results are in agreement with the closed state tightly binding K+ ions by coordinating oxygens of the filter and that the C-termini of the four TM2 helices converge to block the flow of K+ ions. In the open-state, there was rapid-exchange between two conformations of the filter, which were attributed to the low- and high-affinity binding of K+ ions.
KcsA is the largest membrane protein, in terms of mass of the protein (up to 71 kDa) and of the protein-detergent complex (up to ∼130 kDa), for which a structure has been determined by NMR spectroscopy.
Also of interest is that the NMR structure of a water-soluble analogue of KcsA (WSK3) (103 residues, 46 kDa) in H2O/D2O has been determined (Ma et al. Citation2008 and references therein). WSK3 was designed using a statistical approach to predict which residues were likely to be at 35 lipid-exposed positions and to make the protein water-soluble whilst retaining the structure of KcsA. The NMR structure of WSK3 did resemble the crystal structure of KcsA, but new salt bridges and hydrogen bonds were formed, not present in the crystal structure, and there was an altered hydrogen-bond network near the selectivity filter and in the pore.
Proteins with three unique α-helices
The structures of two proteins with three unique transmembrane-spanning α-helices have been determined by solution-state NMR spectroscopy ( and , entries 41 and 42).
Human DAP12-NKG2C immunoreceptor complex
A structure of the heterotrimeric complex formed by association of the transmembrane domains of the DAP12 signalling module with the natural killer cell-activating receptor NKG2C (DAP12-NKG2C) (63 residues, 9.7 kDa) was determined in n-tetradecylphosphocholine (DPC-14)/SDS micelles (Call et al. Citation2010) (). The complex is comprised of two identical DAP12 transmembrane domains (homodimer) and one NKG2C transmembrane domain in which the NKG2C helix packs in an antiparallel orientation along the surface of the DAP12 dimer. This work also determined the structure of the DAP12 transmembrane domain homodimer without the receptor, which was similar to its structure in the heterotrimeric complex. The structure of DAP12 was compared with that of the ζζ transmembrane domain homodimer (see above), which activates the same signalling pathway and has similar rules for association with its partner receptor. A pair of interhelical aspartic acid residues that stabilize the ζζ dimer was found at a different position and with a different side chain conformation in the DAP12 dimer, along with other differences. A threonine residue in the middle of DAP12, not involved in stabilizing the DAP12 dimer, appears to be essential for its assembly with the NKG2C receptor and formation of the heterotrimeric complex. A recent review by Call and Chou (Citation2010) describes the role of solution-state NMR for the structural characterization of membrane-associated protein domains involved in transmembrane signalling.
Figure 9. NMR structure of DAP12-NKG2C complex. (A) Structure of DAP12-NKG2C complex in DPC/SDS and (B) comparison of DAP12 and ζζ TM domain structures. The picture in A was produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005); B is adapted by permission from Macmillan Publishers Ltd: Nature Immunology (Call et al. Citation2010; copyright 2010). This Figure is reproduced in colour in Molecular Membrane Biology online.
Diacylglycerol kinase (DAGK)
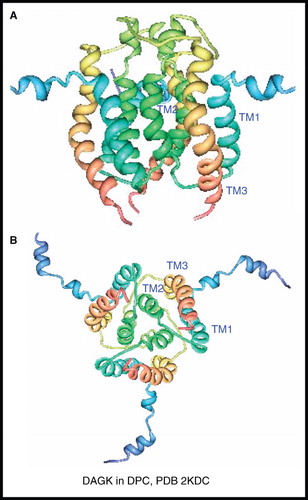
The E. coli integral membrane enzyme diacylglycerol kinase (DAGK) catalyzes the phosphorylation of the lipid diacylglycerol by MgATP to form phosphatidic acid and MgADP. It functions as a homotrimer with each subunit comprised from 121 residues and having three unique transmembrane-spanning α-helices (nine in total, 39 kDa). The backbone NMR resonance assignments for DAGK in DPC micelles (∼ 100 kDa complex at 45°C) using TROSY-based experiments were reported in 2004 (Oxenoid et al. Citation2004). It was not until 2009 that the full three-dimensional NMR structure of the DAGK homotrimer in DPC micelles was published (Van Horn et al. Citation2009) (). The structure shows three-fold symmetry with each subunit having three α-helices in the transmembrane region, as expected, along with two amphipathic helices at the N-terminus (residues 1–25), whose structure could not be precisely determined due to motions in this region. The structure shows that DAGK is domain-swapped, where TM3 packs against the hairpin formed by TM1 and TM2 in the adjacent subunit rather than with TM2 from the same subunit. The authors describe DAGK as resembling ‘a portico’ where TM1 and TM3 act as pillars that terminate at an inner wall formed by TM2 on an adjacent subunit and with a ‘cornice’ on top formed by the loop between TM2 and TM3. Each of the three porticos represent an active site and the results of cysteine-scanning mutagenesis experiments combined with the NMR structure locate DAGK's lipid substrate specificity to the cornice and the overhangs of the cornice are the sites of phosphoryl transfer near the water-membrane interface.
Figure 10. NMR structure of DAGK. Structure of DAGK in DPC; (A) view from the membrane plane and (B) view from the cytosol. These pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
Proteins with four unique α-helices
The structures of four proteins with four unique transmembrane-spanning α-helices have been determined by solution-state NMR spectroscopy ( and , entries 43–47).
Disulphide bond formation enzyme B (DsbB)
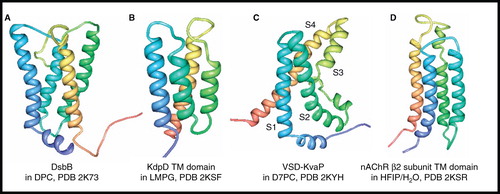
DsbB from E. coli is an enzyme found in the cytoplasmic membrane that is involved in the formation of disulphide bonds in the periplasm. Using membrane-embedded quinones it reoxidizes the reduced form of DsbA, which directly oxidizes periplasmic substrates. The NMR structure of an interloop disulphide bond form of DsbB (183 residues, 21 kDa) was determined in DPC micelles using a combination of NOEs from a deuterated and selectively methyl-protonated sample, backbone dihedral angles from chemical shift analysis, PRE measurements from spin-labelled samples and RDCs from samples aligned in polyacrylamide gels (Zhou et al. Citation2008) (). This is the first NMR structure for a protein with four unique transmembrane-spanning α-helices; it shows the four helices in a left-handed bundle and an N-terminal amphipathic helix (residues 2–9) parallel to the membrane surface. The backbone structure in the transmembrane region is similar to that for DsbB in a crystal structure of the DsbB-DsbA complex determined in n-undecyl-β-D-maltoside/n-nonyl-β-D-thiomaltoside (Inaba et al. Citation2006). Analysis of changes in NMR spectra of DsbB in the presence of DsbA demonstrated two sites for their interaction on the periplasmic loops of DsbB. In a similar way, the binding site of ubiquinone was located at the periplasmic side of DsbB and the structure of the DsbB-ubiquinone complex was consistent with a charge transfer mechanism. The NMR structure of DsbB revealed new information, not obtainable from the crystal structure of the DsbB-DsbA complex, which helped to explain the role of periplasmic loop mobility in electron transfer and the basis for the disulphide exchange pathway in DsbB.
Figure 11. NMR structures of α-helical membrane proteins with four unique α-helices. Structures of (A) DsbB in DPC, (B) KdpD TM domain (397–502) in LMPG, (C) Voltage-Sensor Domain from KvaP (5–147) in D7PC and (D) nAChR β2 subunit TM domain in HFIP/H2O. All pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
Transmembrane domain of histidine kinase receptor KdpD
The backbone structure of the transmembrane domain of the histidine kinase K+ sensor KdpD (105 residues, 11.4 kDa) in LMPG micelles was determined along with those of ArcB and QseC described above (Maslennikov et al. Citation2010). The structure () shows a four-helix bundle in which the second and third helices are relatively short (23–24 Å) compared with helices one and four and with those in ArcB and QseC (30–32 Å). The second helix only interacts with helices one and two, and helices one and four only interact near their cytoplasmic ends, consistent with the loose helix packing observed for all structures in this study.
Voltage-sensor domain of the potassium channel KvAP
Two NMR structural studies were reported in 2010 for the isolated voltage-sensor domain of the potassium channel KvAP (VSD-KvAP) (147 residues, 16.2 kDa) from the hyperthermophilic bacterium Aeropyrum pernix. The first had the VSD solubilized in DPC/LDAO mixed micelles (50 kDa complex at 42°C) (Shenkarev et al. 2010a) and provided structural information that matched well with the crystal structure of the isolated VSD in n-octyl-β-D-glucoside (Jiang et al. Citation2003), which has four transmembrane α-helical segments (S1–S4); it was concluded that this NMR structure of the VSD is in a conformation that corresponds to the open-state of the KvAP channel. The second study determined the structure of the VSD in D7PC (1,2-diheptanoyl-sn-glycero-3-phosphocholine) micelles (50–60 kDa complex at 45°C) (Butterwick and MacKinnon Citation2010) () and was generally in overall agreement with the earlier study and with the crystal structure of the isolated VSD. This NMR structure also showed a 10-residue amphipathic α-helix that precedes S1 and which appears to lie perpendicular to the transmembrane helices, probably interacting with the interfacial region of the D7PC micelle; this helix was not included in the crystal structure. Both NMR studies showed some differences in the S3–S4 paddle region compared with the crystal structure. The NMR and crystal structures both show a significant kink over four residues in the middle of S3 that divides this helix into two separate segments (S3a and S3b), which behave differently during the gating cycle. Both NMR studies also performed backbone dynamics measurements, which were in agreement that different parts of VSD undergo intramolecular motions at several timescales.
Transmembrane domain of nicotinic acetylcholine receptor β2 subunit
The NMR structure of the transmembrane domain of the human nicotinic acetylcholine receptor (nAChR) β2 subunit (164 residues, 18.5 kDa) was determined in a 50% mixture of hexafluoro-isopropanol (HFIP)/H2O (Bondarenko et al. Citation2010) (). The HFIP/H2O mixture was used for solubilization as the β2 transmembrane domain gave poor spectral quality and aggregation in detergents, possibly due to the exposed hydrophobic surface normally occupied by the extracellular domain. The structure revealed a four-helix bundle that was shown to have similarities with the transmembrane domains in structures of the homologous Torpedo nAChR in native membranes (Unwin Citation2005) and the bacterial ion channel GLIC in DDM (Bocquet et al. Citation2009, Hilf and Dutzler Citation2009) determined by cryo-electron microscopy and X-ray crystallography, respectively.
Proteins with seven unique α-helices – sensory rhodopsin II
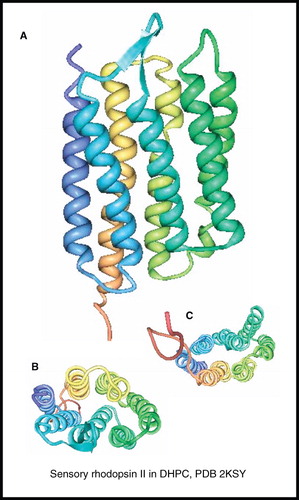
The only membrane protein with seven unique transmembrane-spanning α-helices for which an NMR structure has been determined is the phototaxis receptor sensory rhodopsin II (pSRII) (241 residues, 26.7 kDa) from Natronomonas pharaonis ( and , entry 48). The backbone assignment (> 98%), secondary structure, and dynamics of pSRII in DHPC micelles (70 kDa complex at 50°C) were reported first (Gautier et al. Citation2008), where comparisons with the crystal structure of pSRII in n-octylglucoside (Luecke et al. Citation2001) and backbone assignments (40%) of pSRII in native membranes by solid-state NMR (Etzkorn et al. Citation2007) showed a general overall similarity and differences were attributed to the different sample and temperature conditions that were used. A full three-dimensional NMR structure for pSRII in DHPC micelles was then published (Gautier et al. Citation2010) (), the first for a seven-helix receptor. Complete backbone assignments were made possible by reducing the sample pH from 5.9–5.4, which also made possible the assignments for a nearly complete set of 13Cα and 13Cβ signals. Inter-amide distances (307) were obtained from 15N 3D (nuclear overhauser effect spectroscopy [NOESY]) measurements on a highly deuterated sample and amide-water exchange experiments identified loop regions and solvent-exposed parts of the α-helices. Assignments for almost all methyl groups (140 out of 141) in Ile, Leu and Val residues were achieved from a selectively methyl-protonated sample, which then allowed the measurement of a large number of NOE distances. Further long-range NOE distances were obtained from the methyl groups of Ala, Thr, Met and Ile(γ2) residues in a fully protonated sample; assignment of NOEs involving the side chains of Phe, Tyr and Trp residues were also important. An impressive average number of 15 NOEs per residue were used in calculating the structure. The NMR structure showed the expected seven transmembrane α-helices with the lengths of the helices matching those in the crystal structures and with their packing and orientation also in close agreement.
Figure 12. NMR structure of sensory rhodopsin II. Structure of sensory rhodopsin II in DHPC; (A) view from the membrane plane, (B) view from the cytosol and (C) view from the periplasm. These pictures were produced using the PDB file and PDB Protein Workshop 3.9 (Moreland et al. Citation2005). This Figure is reproduced in colour in Molecular Membrane Biology online.
pSRII is the largest membrane protein, in terms of number of residues and complexity, for which a structure has been determined by NMR spectroscopy. It can be regarded as a model system for GPCRs for which only five unique non-rhodopsin crystal structures have so far been determined (Cherezov et al. Citation2007, Rasmussen et al. Citation2007, Jaakola et al. Citation2008, Warne et al. Citation2008, Chien et al. Citation2010, Wu et al. Citation2010). The strategies used for solving the NMR structure of pSRII should be transferable to GPCRs if sufficient quantities of labelled protein can be obtained and sample conditions can be identified in which the protein is at a sufficient concentration and stable for long enough to acquire NMR spectra that are of sufficient resolution. This may require the construction of thermostable forms of GPCRs in a similar way to that achieved with the β1- and β2-adrenergic receptors (Warne et al. Citation2008, Serrano-Vega and Tate Citation2009) and the adenosine-A2a receptor (Magnani et al. Citation2008).
Backbone resonance assignments have been made for a 24 kDa fragment of the membrane protein TehA with seven out of 10 predicted transmembrane α-helices in LMPG micelles at 40°C (Trbovic et al. Citation2005). The protein was produced by cell-free expression with a yield of 3 mg per millilitre reaction volume. TROSY-based experiments on an 800 MHz instrument with the uniformly-labelled protein allowed unambiguous assignment of 55% of backbone signals; selective labelling with 10 different amino acid types allowed assignment of a further 10% of signals. A combinatorial labelling scheme was then used which achieved 85% assignment. Further assignments or a full structure have yet to be reported for TehA.
Trends for determining the structures of membrane proteins by NMR
Almost all of the structures covered in this review came from proteins that were produced by recombinant expression in E. coli, emphasizing that this is still the system of choice to produce membrane proteins for NMR structural studies. The only exceptions were for some of the studies on the M2 proton channel, which was produced by chemical synthesis, and the work by Maslennikov et al. (Citation2010), which produced transmembrane domains of histidine kinase receptors by cell-free expression.
All of the β-barrel proteins were produced in a denatured and unfolded state before refolding in detergent micelles for NMR sample preparation. A robust refolding protocol provides significant advantages for the structure determination of a membrane protein by NMR including high yields of highly pure protein and high sample reproducibility, which are all important considerations when using costly labelling schemes. The refolding of α-helical membrane proteins from a denatured state has not been so widely successful, though this was achieved in the cases of the M2 proton channel, phospholamban, Ste2P and DAP12-NKG2C.
The range of sample conditions that have been used for determining the NMR structures of membrane proteins shows that there isn't one optimum detergent/lipid that gives the best spectra, but a number of conditions have to be screened on a case by case basis. Deuterated detergent/lipid was used in a majority of cases, which improves spectral quality, as was emphasized by the work on Ste2P. The protein is typically at a concentration of 0.1–2.0 mM in the NMR samples, with an average of ∼1 mM, which is equivalent to 6 mg in 300 μl for a 20 kDa protein. The majority of structures have been determined with the sample at a temperature in the range of ∼25–40°C; higher temperatures were used only with exceptionally stable systems, e.g., 50°C with KcsA and sensory rhodopsin II. The high thermal stability of these proteins clearly contributed to their successful structure determination as they are the largest of all in terms of mass and in terms of number of residues, respectively. Other proteins that were described as being especially stable are all of the β-barrel proteins, the truncated form of MerF and Ste2P. Many of the breakthrough studies were therefore performed with stable systems.
Some of the largest membrane proteins for which NMR structures have been determined function as oligomers, e.g., M2 proton channel (homotetramer), phospholamban (homopentamer), KcsA (homotetramer) and DAGK (homotetramer); this gives them a relatively large mass, but the repeats of transmembrane segments effectively simplifies their spectra, which have fewer signals than a monomeric protein with a comparable overall mass.
Prior to the measurement of restraints for calculating the NMR structure of a membrane protein, its backbone resonances have to be assigned. For the larger systems this generally requires application of TROSY-based multi-dimensional correlation experiments. Side-chain resonances have been successfully assigned by selective proton-labelling of the methyl groups in Ile, Leu and Val residues in a perdeuterated background. Following signal assignment, the type of restraints that have to be measured for structure determination of a moderately large membrane protein include the use of chemical shifts to define secondary structure backbone conformation, 1H-1H NOEs to define the three-dimensional fold of the protein, anisotropic interactions from RDCs in aligned samples, and long-range distances from PRE measurements.
The structural models of the membrane proteins covered in this review have significant differences in their precision and therefore relative resolution. This is apparent by comparing the average number of NMR restraints per residue that were used to calculate the structures (which range from 0.6–36) and the RMSD values for the final structures (which range from 0.3–4 Å) (). If high-resolution structures are considered to have an RMSD of ≤0.4 Å and are based on an average of more than 20 restraints per residue, then the highest-resolution structures are those for Bnip3, ErbB2, EphA1, EphA2, phospholamban, subunit c of F1F0-ATP synthase, Ste2P and sensory rhodopsin II. Many of the RMSD values shown in are for the structured/rigid regions of the protein, such as the transmembrane α-helices, and do not include flexible regions, such as the loops. An impressive exception are the values for the structure of sensory rhodopsin II, which has a backbone RMSD for the transmembrane and loop regions of 0.481 Å and an RMSD of 1.23 Å compared with the 2.1 Å crystal structure of the protein (, entry 48). The inclusion of RDC and PRE restraints provides a significant improvement in the accuracy of NMR structures that can be obtained; for example, refinement of OmpA with 434 RDCs produced a structure with a backbone RMSD for residues in β-strands of 0.48 Å and a backbone RMSD of 1.02 Å compared with the 1.65 Å crystal structure of the protein (, entries 3 and 4).
Conclusions and future perspectives
This review has highlighted an explosion in the number of high-resolution membrane protein structures determined by NMR spectroscopy over the past few years, especially for more complex α-helical systems. This has been made possible through obtaining sufficient quantities of highly pure and stable protein, the development of new labelling schemes and NMR experiments, the application of novel strategies for signal assignment and structure calculation, and the availability of higher-field NMR magnets and cryogenically-cooled NMR probes. The major limiting factor behind obtaining further structures of the larger α-helical membrane proteins (with seven transmembrane helices or more) is obtaining sufficient quantities of labelled and purified protein that is stable for long periods at elevated temperatures, and obtaining well-dispersed and high-sensitivity NMR spectra. Further advances in systems for membrane protein expression and labelling and in NMR methodology will make the structure-determination of such systems more routine.
The types of α-helical membrane proteins with huge potential for applying NMR structural studies to better understand their molecular mechanisms and roles in biological processes and for providing information to assist drug design, include histidine and tyrosine kinase receptors, GPCRs (Tapaneeyakorn et al. Citation2011) and transport proteins. No crystal structure has yet been determined for a full-length histidine kinase receptor, which are targets for developing novel antibiotics (Casino et al. Citation2010). The work on ArcB, QseC and KdpD has demonstrated that if sufficient quantities of labelled transmembrane domains for a histidine kinase receptor can be obtained, then its NMR structure can be determined. No NMR structure has been reported for a full-length human GPCR; however, the work with sensory rhodopsin II has demonstrated that it is feasible to obtain the structure of a seven-transmembrane spanning α-helical protein by NMR and the methods used should be transferable to human GPCRs if sufficient quantities of labelled and thermally-stable protein can be obtained. This will require further advances in heterologous expression systems and in the design and construction of thermostable GPCRs. No NMR structure has yet been reported for a transport protein, which can have up to 14 transmembrane-spanning α-helices. Advances in all areas of sample production and NMR methodology will likely make this possible in the not too distant future, indeed our own work has shown that it is possible to obtain high-resolution solution-state TROSY spectra from selectively-labelled and detergent-solubilized samples of a 12-helix transport protein (Patching, Kalverda, Henderson, Homans, unpublished work). Rather than working on full-length proteins, a reductionist approach is still likely to be used in the short term with many of the more challenging systems, be it NMR structural studies on just transmembrane domains, as with ArcB, QseC and KdpD or fragments of GPCRs/receptors, as with Ste2P and nAChR β2 subunit.
The amino acid-selective isotopic labelling of larger systems to simplify spectra and to assist signal assignment can be achieved in E. coli by including labelled amino acids or their precursors in the growth medium and/or by using auxotrophic host strains (Ou et al. Citation2001, Patching et al. Citation2008a). Combinatorial selective labelling schemes can be used with E. coli and with cell-free expression systems, for example, by detecting unique pairs using 15N/13C and 15N/14N labelling patterns (Parker et al. Citation2004, Craven et al. Citation2007), the combinatorial dual-labelling (CDL) strategy developed by Masslenikov et al. 2010, stereo-array isotope labelling (SAIL) (Kainosho et al. Citation2006, Takeda et al. Citation2010), the use of [1,3-13C]- and [2-13C]glycerol (Higman et al. Citation2009) and unlabelling (or reverse labelling) schemes (Etzkorn et al. Citation2007) for solid-state NMR and by applying computer algorithms, such as the recently developed UPLABEL (Hefke et al. Citation2011) for optimizing labelling schemes. The benefits of performing ILV methyl-group labelling have been discussed earlier in this review. Segmental isotope labelling (Skrisovska et al. Citation2010) may also provide an important contribution. The selective labelling of eukaryotic proteins by expression systems that use insect and mammalian cells is more challenging, however.
Cell-free expression is likely to make an important contribution for the future structure-determination of the more complex α-helical membrane proteins by NMR spectroscopy. Cell-free systems allow the relatively quick production of milligram quantities of highly pure protein directly into a membrane-mimetic combined with isotopic labelling at any positions with little or no scrambling (Reckel et al. Citation2010, Sobhanifar et al. Citation2010) and there have been recent advances in adapting the systems to suit the requirements of individual membrane proteins (Junge et al. Citation2011).
Care has to be taken when using detergent micelles for solubilization of membrane proteins for structural studies as these are not the best mimic for a lipid membrane, even if they do generally provide the highest resolution NMR spectra. A developing area is the use of reverse micelles (Kielec et al. Citation2009, Citation2010) and non-micellar systems to solubilize membrane proteins for solution-state NMR structural studies. Amphipols, lipid bilayer systems such as bicelles and nanolipoprotein particles (NLPs) and fluorinated surfactants all provide an environment for the protein that is nearer to native membrane conditions than do detergent micelles (Popot Citation2010, Raschle et al. Citation2010). Nanodiscs (Glück et al. Citation2009) and styrene maleic acid lipid particle (SMALP) systems have also been recently combined with cell-free expression to produce functional membrane proteins for NMR studies (Shenkarev et al. 2010b, Rajesh et al. Citation2011).
The continuous development of new solution-state NMR pulse sequences and the availability of larger NMR magnets and of higher sensitivity probes will also contribute to achieving structures of the larger α-helical membrane proteins; for example, using direct carbon (13C) detection and fast methods (Bermel et al. Citation2008, Citation2009, Felli and Brutscher Citation2009) combined with cryogenically-cooled carbon probes.
Compared with solution-state NMR spectroscopy, solid-state NMR spectroscopy is still a developing technique and its full potential for structure-function studies on membrane proteins is yet to be realized. As there is no limit to the size of the complex that can be studied by solid-state NMR it allows experiments on membrane proteins expressed in their native membranes or reconstituted in lipid systems that closely mimic their native membranes; there is therefore more confidence in the physiological relevance of structural information obtained under these conditions. The experiments are often performed at temperatures below 0°C, so sample stability is not usually a major concern. Although it is not yet possible to routinely determine the complete structures of membrane proteins by solid-state NMR, the technique is uniquely able to determine the structures of bound ligands and their binding sites with membrane proteins, as demonstrated by the work on the M2 proton channel, and it can be used to investigate other structural features and mechanistic properties of the proteins. A comprehensive description of solid-state NMR studies on membrane proteins is beyond the scope of this review, but there has been recent work relating to the already mentioned proteins. Experiments on OmpX in magnetically-oriented phospholipid bilayers has shown that it adopts a 7° tilt of its β-barrel axis relative to the membrane normal (Mahalakshmi and Marassi Citation2008). Proton-detected 2D H/N solid-state NMR spectra have been obtained for perdeuterated and partially proton-back-substituted samples of OmpG in deuterated lipids and compared with solution-state TROSY spectra of OmpG in DDM micelles (Linser et al. Citation2011). The conformation of the VDAC1 N-terminus in lipid membranes has been elucidated (Schneider et al. Citation2010). Conformational dynamics in response to potassium ion concentration have been investigated in the selectivity filter of KcsA (Varga et al. Citation2007, Bhate et al. Citation2010). Solid-state NMR combined with neutron diffraction has been used to investigate the structure and hydration of the VSD from KvaP in native membranes, showing how the protein influences the structure of the bilayer (Krepkiy et al. Citation2009), and the orientation and depth of insertion of the S4 helix from the VSD in lipid bilayers has also been investigated (Doherty et al. Citation2010). Work on the nAChR includes determining the conformation of acetylcholine bound to the protein in native membranes (Williamson et al. Citation2007), determination of the high-resolution structure of the putative hinge region in M2 channel-lining segments of the protein (Long et al. Citation2007), and identification of the binding site for short neurotoxin II (Krabben et al. Citation2009). Time-resolved 31P magic-angle spinning (MAS) NMR has been used to simultaneously and directly detect both ATP hydrolysis and diacylglycerol phosphorylation by membrane-bound DAGK and to investigate the kinetics of the system (Ullrich et al. Citation2011). Chemical shift assignments have been made for DsbB by 3D MAS NMR (Li et al. Citation2007, Citation2008) and the charge-transfer complex between ubiquinone-8 and DsbB has been investigated (Tang et al. Citation2011). Complex formation and light activation have been investigated with sensory rhodopsin II and its transducer in native-like membranes (Etzkorn et al. Citation2010) and the conformation of a 7-TM sensory rhodopsin in lipids has been characterized (Shi et al. Citation2011). Solid-state NMR can also be used to investigate the specificity and properties of ligand binding to wild-type and mutant forms of membrane proteins (Patching et al. Citation2004a, Citation2008b, Xie et al. Citation2004), including the measurement of binding constants and rate constants for dissociation with weakly-binding (μM-mM affinity) ligands (Patching et al. Citation2004b). All of this type of information can be combined with structures from crystallography and/or solution-state NMR to understand better how native ligands or drug molecules interact with membrane proteins and can be used to assist the design of new treatments for human disease. Further developments in labelling schemes, pulse sequences and probe design for solid-state NMR will move it closer to determining the full structures of membrane proteins. For example, the potential impact of sensitivity enhancements that can be achieved by performing NMR experiments with the sample at cryogenic temperatures, as developed by the groups of Levitt, Samoson and Tycko (Carravetta et al. Citation2006, Thurber and Tycko Citation2008) is yet to be shown for membrane proteins.
This review has demonstrated a recent explosion in the number of membrane protein structures determined by NMR spectroscopy, especially those with multiple transmembrane-spanning α-helices. This has come as a result of new expression systems and strategies for isotopic labelling, sample preparation, signal assignment and structure calculation combined with developments in NMR pulse sequences and probe design and higher-field magnets. The next 5–10 years are likely to see many more NMR structures from the huge number of membrane proteins yet to be characterized. The structures obtained so far have also highlighted the benefits of combining information from solution- and solid-state NMR spectroscopy with that from other biophysical and biochemical techniques for understanding the structure and function of membrane proteins.
Acknowledgements
The author thanks Peter Henderson and Steve Homans (University of Leeds) and Malcolm Levitt (University of Southampton) for their support.
Declaration of interest: This work was funded by grants from the EPSRC (EP/G035695/1) and EU (201924, EDICT).The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Akermoun M, Koglin M, Zvalova-Iooss D, Folschweiller N, Dowell SJ, Gearing KL. 2005. Characterization of 16 human G protein-coupled receptors expressed in baculovirus-infected insect cells. Protein Expres Purif 44:65–74.
- Arinaminpathy Y, Khurana E, Engelman DE, Gerstein MB. 2009. Computational analysis of membrane proteins: The largest class of drug targets. Drug Discov Today 14:1130–1135.
- Arora A, Abildgaard F, Bushweller JH, Tamm LK. 2001. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struct Biol 8:334–338.
- Bader R, Lerch M, Zerbe O. 2002. NMR of membrane-associated peptides and proteins. In: Zerbe O, editor. BioNMR in drug research. Weinheim: Wiley-VCH Verlag GmbH and Co. pp 95–120.
- Baker KA, Tzitzilonis C, Kwiatkowski W, Choe S, Riek R. 2007. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat Struct Biol 14:1089–1095.
- Bayrhuber M, Meins T, Habeck M, Becker S, Giller K Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K. 2008. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci USA 105:15370–15375.
- Bermel W, Felli IC, Kummerle R, Pierattelli R. 2008. C-13 direct-detection biomolecular NMR. Concept Magnetic Res 32A:183–190.
- Bermel W, Bertini I, Felli IC, Pierattelli R. 2009. Speeding up 13C direct detection biomolecular NMR spectroscopy. J Am Chem Soc 131:15339–15345.
- Bhate MP, Wylie BJ, Tian L, McDermott AE. 2010. Conformational dynamics in the selectivity filter of KcsA in response to potassium ion concentration. J Mol Biol 401:155–166.
- Bocharov EV, Pustovalova YE, Pavlov KV, Volynsky PE, Goncharuk MV, Ermolyuk YS, Karpunin DV, Schulga AA, Kirpichnikov MP, Efremov RG, Maslennikov IV, Arseniev AS. 2007. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilisation as a cell death trigger. J Biol Chem 282:16256–16266.
- Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, Arseniev AS. 2008a. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem 283:6950–6956.
- Bocharov EV, Mayzel ML, Volynsky PE, Goncharuk MV, Ermolyuk YS, Schulga AA, Artemenko EO, Efremov, RG, Arseniev AS. 2008b. Spatial structure and pH-dependent conformational diversity of dimeric transmembrane of the receptor tyrosine kinase EphA1. J Biol Chem 283:29385–29395.
- Bocharov EV, Mayzel ML, Volynsky PE, Mineev KS, Tkach EN, Ermolyuk YS, Schulga AA, Efremov, RG, Arseniev AS. 2010. Left-handed dimer of EphA2 transmembrane domain: Helix packing diversity among receptor tyrosine kinases. Biophys J 98:881–889.
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux J-P, Delarue M, Corringer P-J. 2009. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457:111–114.
- Bondarenko V, Tillman T, Xu Y, Tang P. 2010. NMR structure of the transmembrane domain of the N-acetylcholine receptor β2 subunit. BBA-Biomembranes 1798:1608–1614.
- Butterwick JA, MacKinnon R. 2010. Solution structure and phosholipid interactions of the isolated voltage-sensor domain from KvAP. J Mol Biol 403:591–606.
- Cady SD, Mishanina TV, Hong M. 2009. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: The role of Ser31 in amantadine binding. J Mol Biol 385:1127–1141.
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. 2010. Structure of the amantadine binding site of influenza proton channels in lipid bilayers. Nature 463:689–692.
- Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, Wucherpfennig KW. 2006. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127:355–368.
- Call ME, Wucherpfennig KW, Chou JJ. 2010. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol 11:1023–1029.
- Call ME, Chou JJ. 2010. A view into the blind spot: Solution NMR provides new insights into signal transduction across the lipid bilayer. Structure 18:1559–1569.
- Carravetta M, Johannessen OG, Levitt MH, Heinmaa I, Stern R, Samoson A, Horsewill AJ, Murata Y, Komatsu K. 2006. Cryogenic NMR spectroscopy of endohedral hydrogen-fullerene complexes. J Chem Phys 124:104507.
- Casino P, Rubio V, Marina A. 2010. The mechanism of signal transduction by two-component systems. Curr Opin Struc Biol 20:1–9.
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. 2007. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318:1258–1265.
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. 2010. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095.
- Chill JH, Louis JM, Miller C, Bax A. 2006. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Prot Sci 15:684–698.
- Cierpicki T, Liang B, Tamm LK, Bushweller BH. 2006. Increasing the accuracy of solution NMR structures of membrane proteins by application of residual dipolar couplings. High resolution structure of outer membrane protein A. J Am Chem Soc 128:6947–6951.
- Congreve M, Marshall F. 2010. The impact of GPCR structures on pharmacology and structure-based drug design. Brit J Pharmacol 159:986–996.
- Craven CJ, Al-Owais M, Parker, MJ. 2007. A systematic analysis of backbone amide assignments achieved via combinatorial selective labelling of amino acids. J Biomol NMR 38:151–159.
- Czabotar PE, Martin SR, Hay AJ. 2004. Studies of structural changes in the M2 proton channel of influenza A virus by tryptophan fluorescence. Virus Res 99:57–61.
- De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. 2006. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc 128:12256–12267.
- De Angelis AA, Opella SJ. 2007. Bicelle samples for solid-state NMR of membrane proteins. Nature Protocols 2:2332–2338.
- Doherty T, Su Y, Hong M. 2010. High-resolution orientation and depth of insertion of the voltage-sensing S4 helix of a potassium channel in lipid bilayers. J Mol Biol 401:642–652.
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. 1998. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 280:69–77.
- Drews J. 2000. Drug discovery: A historical perspective. Science 287:1960–1964.
- Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. 2007. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew Chem Int Edit 46:459–462.
- Etzkorn M, Seidel K, Li L, Martell S, Geyer M, Engelhard M, Baldus M. 2010. Complex formation and light activation in membrane-embedded sensory rhodopsin II as seen by solid-state NMR spectroscopy. Structure 18:293–300.
- Fagerberg L, Jonasson K, von Heijne G, Uhlen M, Berglund L. 2010. Prediction of the human membrane proteome. Proteomics 10:1141–1149.
- Felli IC, Brutscher B. 2009. Recent advances in solution NMR: Fast methods and heteronuclear direct detection. Chem Phys Chem 10:1356–1368.
- Fernández C, Adeishvili K, Wüthrich K. 2001. Transverse relaxation-optimised spectroscopy with the outer membrane protein OmpX in dihexanoyl phosphatidylcholine micelles. Proc Natl Acad Sci USA 98:2358–2363.
- Fernández C, Hilty C, Wider G, Güntert P, Wüthrich K. 2004. NMR structure of the integral membrane protein OmpX. J Mol Biol 336:1211–1221.
- Gautier A, Kirkpatrick JP, Nietlispach D. 2008. Solution-state NMR spectroscopy of a seven-helix transmembrane protein receptor: Backbone assignment, secondary structure, and dynamics. Angew Chem Int Ed 47:7297–7300.
- Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. 2010. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol 17:768–775.
- Girvin ME, Rastogi VK, Abildgaard F, Markley JL, Fillingame RH. 1998. Solution structure of the transmembrane H+-transporting subunit c of the F1F0 ATP synthase. Biochemistry 37:8817–8824.
- Glück JM, Wittlich M, Feuerstein S, Hoffmann S, Willbold D, Koenig BW. 2009. Integral membrane proteins in nanodiscs can be studied by solution NMR. J Am Chem Soc 131:12060–12061.
- Gullion T, Schaefer J. 1989a. Rotational-echo double-resonance NMR. J Magn Reson 81:196–200.
- Gullion T, Schaefer J. 1989b. Detection of weak heteronuclear dipolar coupling by rotational-echo double-resonance NMR. Adv Magn Reson 13:57–83.
- Hays FA, Roe-Zurz Z, Stroud RM. 2010. Overexpression and purification of integral membrane proteins in yeast. Methods Enzymol 470:695–707.
- Hefke F, Bagaria A, Reckel S, Ullrich SJ, Dötsch V, Glaubitz C, Güntert P. 2011. Optimization of amino acid type-specific (13)C and (15)N labeling for the backbone assignment of membrane proteins by solution- and solid-state NMR with the UPLABEL algorithm. J Biomol NMR 48:75–84.
- Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, van Rossum BJ, Oschkinat H. 2009. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively C-13-labelled proteins. J Biomol NMR 44:245–260.
- Hilf RJ, Dutzler R. 2009. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457:115–118.
- Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. 2008. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321:1206–1210.
- Hiller S, Wagner G. 2009. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Curr Opin Struc Biol 19:396–401.
- Hopkins AL, Groom CR. 2002. The druggable genome. Nat Rev Drug Discov 1:727–730.
- Howell SC, Mesleh MF, Opella SJ. 2005. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry 44:5196–5206.
- Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. 2007. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys J 92:4335–4343.
- Hu F, Luo W, Hong M. 2010. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330:505–508.
- Hwang PM, Choy W-Y, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Prive GG, Bishop RE, Kay LE. 2002. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Natl Acad Sci USA 99:13560–13565.
- Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. 2006. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulphide bond generation. Cell 127:789–801.
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. 2008. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217.
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. 2003. X-ray crystal structure of a voltage-dependent K+ channel. Nature 423:33–41.
- Johansson MU, Alioth S, Hu K, Walser R, Koebnik R, Pervushin K. 2007. A minimal transmembrane beta-barrel platform protein studied by nuclear magnetic resonance. Biochemistry 46:1128–1140.
- Junge F, Haberstock S, Roos C, Stefer S, Proverbio D, Dötsch V, Bernhard F. 2011. Advances in cell-free protein synthesis for the functional and structural analysis of membrane proteins. New Biotechnol 28:262–271.
- Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Ono AM, Güntert P. 2006. Optimal isotope labelling for NMR protein structure determinations. Nature 440:52–57.
- Kielec JM, Valentine KG, Babu CR, Wand AJ. 2009. Reverse micelles in integral membrane protein structural biology by solution NMR spectroscopy. Structure 17:345–351.
- Kielec JM, Valentine KG, Wand AJ. 2010. A method for solution NMR structural studies of large integral membrane proteins: Reverse micelle encapsulation. BBA-Biomembranes 1798:150–160.
- Kim H-J, Howell SC, Van Horn, WD, Ho Jeon Y, Sanders CR. 2009. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Mag Res Sp 55:335–360.
- Koth CMM, Payandeh J. 2009. Strategies for the cloning and expression of membrane proteins. Adv Protein Chem Struct Biol 76:43–86.
- Krabben L, van Rossum B-J, Jehle S, Bocharov E, Lyukmanova EN, Schulga AA, Arseniev A, Hucho F, Oschkinat H. 2009. Loop 3 of short neurotoxin II is an additional interaction site with membrane-bound nicotinic acetylcholine receptor as detected by solid-state NMR spectroscopy. J Mol Biol 390:662–671.
- Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, Swartz KJ. 2009. Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462:473–479.
- Krueger-Koplin RD, Sorgen PL, Kruger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich TA, Girvin, ME. 2004. An evaluation of detergents for NMR structural studies of membrane proteins. J Biomol NMR 17:43–57.
- Largerström MC, Schiöth HB. 2008. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357.
- Lau T-L, Kim C, Ginsberg MH, Ulmer TS. 2009. The structure of the integrin αIIbβ3 transmembrane complex explains integrin transmembrane signalling. EMBO J 28:1351–1361.
- Lee D, Hilty C, Wider G, Wüthrich K. 2006. Effective rotational correlation times of proteins from NMR relaxation interference. J Magn Reson 178:72–76.
- Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. 2007. Partial 13C and 15N chemical-shift assignmants of the disulphide-bond-forming enzyme DsbB by 3D magic-angle spinning NMR spectroscopy. ChemBioChem 8:434–442.
- Li Y, Berthold DA, Gennis RB, Rienstra CM. 2008. Chemical shift assignment of the transmembrane helices of DsbB, a 20-kDa integral membrane enzyme, by 3D magic-angle spinning NMR spectroscopy. Protein Sci 17:199–204.
- Liang B, Bushweller JH, Tamm LK. 2006. Site-directed parallel spin-labelling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc 128:6947–6951.
- Liang B, Tamm LK. 2007. Structure of outer membrane protein G by solution NMR spectroscopy. Proc Natl Acad Sci USA 104:16140–16145.
- Linser R, Dasari M, Hiller M, Higman V, Fink U, Lopez del Amo J-M, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, Reif B. 2011. Proton-detected solid-state NMR spectroscopy of fibrillar and membrane proteins. Angew Chem Int Ed 50:4508–4512.
- Liu JF, Rost B. 2001. Comparing function and structure between entire proteomes. Prot Sci 10:1970–1979.
- Long JR, Mills FD, Raucci F. 2007. A high-resolution structure of the putative hinge region in M2 channel-lining segments of the nicotinic acetylcholine receptor. BBA-Biomembranes 1768:2961–2970.
- Lorch M, Faham S, Kaiser C, Weber I, Mason AJ, Bowie JU, Glaubitz C. 2005. How to prepare membrane proteins for solid-state NMR: A case study on the alpha-helical integral membrane protein diacylglycerol kinase from E. coli. Chembiochem 6:1693–1700.
- Luecke H, Schobert B, Lanyi JK, Spudich EN, Spudich JL. 2001. Crystal structure of sensory rhodopsin II at 2.4 angstroms: insights into colour tuning and transducer interaction. Science 293:1499–1503.
- Lundstrom K. 2006. Latest development in drug discovery on G protein-coupled receptors. Curr Protein Pept Sci 7:465–470.
- Ma D, Tillman TS, Tang P, Meirovitch E, Eckenhoff R, Carnini A, Xu Y. 2008. NMR studies of a channel protein without membranes: Structure and dynamics of water-solubilised KcsA. Proc Natl Acad Sci USA 105:16537–16542.
- MacKenzie KR, Prestegard JH, Engelman DM. 1997. A transmembrane helix dimer: Structure and implications. Science 276:131–133.
- Magnani F, Shibata Y, Serrano-Vega MJ, Tate CG. 2008. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc Natl Acad Sci USA 105:10744–10749.
- Mahalakshmi R, Marassi FM. 2008. Orientation of the Escherichia coli outer membrane protein OmpX in phospholipid bilyaer membranes determined by solid-state NMR. Biochemistry 47:6531–6538.
- Maslennikov I, Klammt C, Hwang E, Kefala G, Okamura M, Esquivies L, Mors K, Glaubitz C, Kwiatkowski W, Jeon YH, Choe S. 2010. Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc Natl Acad Sci USA 107:10902–10907.
- Midgett CR, Madden DR. 2007. Breaking the bottleneck: eukaryotic membrane expression for high-resolution structural studies. J Struct Biol 160:265–274.
- Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV, Arseniev AS. 2010. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J Mol Biol 400:231–243.
- Moreland JL, Gramada A, Buzko1 OV, Zhang Q, Bourne PE. 2005. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinformatics 6:21.
- Müller DJ, Wu N, Palczewski K. 2008. Vertebrate membrane proteins: Structure, function and insights from biophysical approaches. Pharmacol Rev 60:43–78.
- Neumoin A, Cohen LS, Arshava B, Tantry S, Becker JM, Zerbe O, Naider F. 2009. Structure of a double transmembrane fragment of a G-protein-coupled receptor in micelles. Biophys J 96:3187–3196.
- Ou HD, Lai HC, Serber Z, Dötsch V. 2001. Efficient identification of amino acid types for fast protein backbone assignments. J Biomol NMR 21:269–273.
- Overington JP, Al-Lazikani B, Hopkins AL. 2006. Opinion – how many drug targets are there? Nat Rev Drug Discov 5:993–996.
- Oxenoid K, Kim HJ, Jacob J, Sönnichsen FD, Sanders CR. 2004. NMR assignments for a helical 40 kDa membrane protein. J Am Chem Soc 126:5048–5049.
- Oxenoid K, Chou JJ. 2005. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci USA 102:10870–10875.
- Page RC, Moore JD, Nguyen HB, Sharma M, Chase R, Philip Gao F, Mobley CK, Sanders CR, Ma L, Sönnichsen FD, Lee S, Howell SC, Opella SJ, Cross TA. 2006. Comprehensive evaluation of solution nuclear magnetic resonance spectroscopy sample preparation for helical integral membrane proteins. J Struct Func Genom 7:51–64.
- Parker MJ, Aulton-Jones M, Hounslow AM, Craven CJ. 2004. A combinatorial selective labelling method for the assignment of backbone amide NMR resonances. J Am Chem Soc 126:5020–5021.
- Patching SG, Herbert RB, O'Reilly J, Brough AR, Henderson PJF. 2004a. Low (13)C-background for NMR-based studies of ligand binding using (13)C-depleted glucose as carbon source for microbial growth: (13)C-Labeled glucose and (13)C-forskolin binding to the galactose-H(+) symport protein GalP in Escherichia coli. J Am Chem Soc 126:86–87.
- Patching SG, Brough AR, Herbert RB, Rajakarier AR, Henderson PJF, Middleton DA. 2004b. Substrate affinities for membrane transport proteins determined by 13C cross-polarization magic-angle spinning nuclear magnetic resonance spectroscopy. J Am Chem Soc 126:3072–3080.
- Patching SG, Henderson PJF, Herbert RB, Middleton DA. 2008a. Solid-state NMR spectroscopy detects interactions between tryptophan residues of the E. coli sugar transporter GalP and the alpha-anomer of the D-glucose substrate. J Am Chem Soc 130:1236–1244.
- Patching SG, Psakis G, Baldwin SA, Baldwin J, Henderson PJF, Middleton DA. 2008b. Relative substrate affinities of wild-type and mutant forms of the Escherichia coli sugar transporter GalP determined by solid-state NMR. Mol Memb Biol 25:474–484.
- Pautsch A, Schulz GE. 1998. Structure of the outer membrane protein A transmembrane domain. Nat Struct Biol 5:1013–1017.
- Pervushin K, Riek R, Wider K, Wüthrich K. 1997. Attenuated T-2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371.
- Pielak RM, Schnell JR, Chou JJ. 2009. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA 106:7379–7384.
- Pielak RF, Chou JJ. 2010. Solution NMR structure of the V27A drug resistant mutant of influenza A channel. Biochem Biophys Res Commun 401:58–63.
- Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA 94:11301–11306.
- Poget SF, Girvin ME. 2007. Solution NMR of membrane proteins in bilayer mimics: Small is beautiful, but sometimes bigger is better. BBA-Biomembranes 1768:3098–3106.
- Popot J-L. 2010. Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem 79:737–775.
- Rajesh S, Knowles T, Overduin M. 2011. Production of membrane proteins without cells or detergents. New Biotechnol 28:250–254.
- Raschle T, Hiller S, Etzkorn M, Wagner G. 2010. Nonmicellar systems for solution NMR spectrocopy of membrane proteins. Curr Opin Struc Biol 20:471–479.
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti R.F, Schertler GF, Weis WI, Kobilka BK. 2007. Crystal structure of the human β2-adrenergic G-protein-coupled receptor. Nature 450:383–387.
- Reckel S, Sobhanifar S, Durst F, Löhr F, Shirokov VA, Dötsch V, Bernhard F. 2010. Strategies for the cell-free expression of membrane proteins. Methods Mol Biol 607:187–212.
- Renault M, Saurel O, Czaplicki J, Demange P, Gervais V, Löhr F, Reat V, Piotto M, Milon A. 2009. Solution state NMR structure and dynamics of KpOmpA, a 210 residue transmembrane domain possessing a high potential for immunological applications. J Mol Biol 385:117–130.
- Rosenbaum DM, Rasmussen SGF, Kobilka BK. 2009. The structure and function of G-protein-coupled receptors. Nature 459:356–363.
- Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K. 1998. TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA 95:13585–13590.
- Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K. 1999. TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J Am Chem Soc 121:844–848.
- Sanders CR, Sönnichsen FD. 2006. Solution NMR of membrane proteins: Practice and challenges. Magn Reson Chem 44:S24–40.
- Schneider R, Etzkorn M, Giller K, Daebel V, Eisfeld Jörg, Zweckstetter M, Griesinger C, Becker S, Lange A. 2010. The native conformation of the human VDAC1 N terminus. Angew Chem Int Edit 49:1882–1885.
- Schnell JR, Chou JJ. 2008. Structure and mechanism of the M2 proton channel of inflenza A virus. Nature 451:591–595.
- Serrano-Vega MJ, Tate CG. 2009. Transferability of thermostabilizing mutations between beta-adrenergic receptors. Mol Memb Biol 26:385–396.
- Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou H-X, Cross TA. 2010. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 330:509–512.
- Shenkarev ZO, Paramonov AS, Lyukmanova EN, Shingarova LN, Yakimov SA, Dubinnyi MA, Chupin VV, Kirpichnikov MP, Blommers MJJ, Arseniev AS. 2010a. NMR structural and dynamical investigation of the isolated voltage-sensing domain of the potassium channel KvAP: Implications for voltage gating. J Am Chem Soc 132:5630–5637.
- Shenkarev ZO, Lyukmanova EN, Paramonov AS, Shingarova LN, Chupin VV, Kirpichnikov, MP, Blommers MJ, Arseniev AS. 2010b. Lipid-protein nanodiscs as reference medium in detergent screening for high-resolution NMR studies of integral membrane proteins. J Am Chem Soc 132:5628–5629.
- Shi L, Kawamura I, Jung K-H, Brown LS, Ladizhansky. 2011. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew Chem Int Edit 50:1302–1305.
- Skrisovska L, Schubert M, Allain FH-T. 2010. Recent advances in segmental isotope labelling of proteins: NMR applications to large proteins and glycoproteins. J Biomol NMR 46:51–65.
- Sobhanifar S, Reckel S, Junge F, Schwarz D, Kai L, Karbyshev M, Löhr F, Bernhard F, Dötsch V. 2010. Cell-free expression and stable isotope labelling strategies for membrane proteins. J Biomol NMR 46:33–43.
- Stouffer AL, Acharya R, Salom D, Levine AS, Costanzo LD, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. 2008. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 451:596–599.
- Subbarao GV, van den Berg G. 2006. Crystal structure of the monomeric porin OmpG. J Mol Biol 360:750–759.
- Sulistijo ES, MacKenzie KR. 2009. Structural basis for dimerisation of the BNip3 transmembrane domain. Biochemistry 48:5106–5120.
- Takeda M, Ono AM, Terauchi T, Kainosho M. 2010. Application of SAIL phenylalanine and tyrosine with alternative isotope-labelling patterns for protein structure determination. J Biomol NMR 46:45–49.
- Takegoshi K, Kakamura S, Teraoa T. 2001. 1H-13C dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett 344:631–637.
- Takeuchi K, Takahashi H, Kewano S, Shimada I. 2007. Identification and characterisation of the slowly exchanging pH-dependent conformational rearrangement in KcsA. J Biol Chem 282:15179–15186.
- Tamm LK, Liang B. 2006. NMR of membrane proteins in solution. Prog Nucl Mag Res Sp 48:201–210.
- Tang Y, Zaitseva F, Lamb RA, Pinto LH. 2002. The gate of the influenza virus proton channel is formed by a single tryptophan residue. J Biol Chem 277:39880–39886.
- Tang M, Sperling LJ, Berthold DA, Nesbitt AE, Gennis RB, Rienstra CM. 2011. Solid-state NMR study of the charge-transfer complex between ubiquinone-8 and disulfide bond generating membrane protein DsbB. J Am Chem Soc 133: 4359–4366.
- Tapaneeyakorn S, Goddard AD, Oates J, Willis CL, Watts A. 2011. Solution- and solid-state NMR studies of GPCRs and their ligands. BBA-Biomembranes 1808:1462–1475.
- Thurber KR, Tycko R. 2008. Biomolecular solid state NMR with magic-angle spinning at 25 K. J Magn Reson 195:179–186.
- Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. 2009. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci USA 106:10165–10170.
- Trbovic N, Klammt C, Koglin A, Löhr F, Bernhard F, Dötsch V. 2005. Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc 127:13504–13505.
- Tugarinov V, Kay LE. 2003. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labelling strategy and novel NMR methods. J Am Chem Soc 125:13868–13878.
- Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J. 2008. The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolic gating. Proc Natl Acad Sci USA 105:17742–17747.
- Ullrich SJ, Hellmich UA, Ullrich S, Glaubitz C. 2011. Interfacial enzyme kinetics of a membrane bound kinase analyzed by real-time MAS-NMR. Nat Chem Biol 7:263–270.
- Unwin N. 2005. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol 346:967–989.
- Van Horn WD, Kim H-J, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sönnichsen FD, Sanders CR. 2009. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science 324:1726–1729.
- Varga K, Tian L, McDermott AE. 2007. Solid-state NMR study and assignments of the KcsA potassium ion channel of S. lividans. BBA-Biomembranes 1774:1604–1613.
- Vogt J, Schulz GE. 1999. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure 7:1301–1309.
- von Heijne G. 2007. The membrane protein universe: what's out there and why bother? J Int Med 261:543–557.
- Wallin E, von Heijne G. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Prot Sci 7:1029–1038.
- Wang C, Takeuchi K, Pinto LH, Lamb RA. 1993. Ion channel activity of influenza A virus M2 protein: characterisation of the amantadine block. J Virol 67:5585–5594.
- Wang J, Pielak RM, McClintock MA, Chou JJ. 2009. Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol 16:1267–1271.
- Wang J, Qiu JX, Soto C, DeGrado WF. 2011. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr Opin Struc Biol 21:68–80.
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. 2008. Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454:486–U2.
- Watanabe H, Koopmann TT, Scouarnec SL, Yang T, Ingram CR, Schott J-J, Demolombe S, Probust V, Anselme F, Escande D, Wiesfeld ACP, Pfuefer A, Kääb S, Wichmann H-E, Hasdemir C, Aizawa Y, Wilde AAM, Roden DM, Bezzina CR. 2008. Sodium channel β1subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118:2260–2268.
- Watts A. 2007. Solid state NMR for studying membrane proteins. Supermolec Struct Funct 9:45–73.
- White SH. 2009. Biophysical dissection of membrane proteins. Nature 459:344–346.
- Williamson PTF, Verhoeven A, Miller KW, Meier BH, Watts A. 2007. The conformation of acetycholine at its target site in the membrane-embedded nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 104:18031–18036.
- Williamson PTF. 2009. Solid-state NMR for the analysis of high-affinity ligand/receptor interactions. Concept Magnetic Res 34A:144–172.
- Wu CH, Ramamoorthy A, Opella SJ. 1994. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson 109:270–272.
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel, TM, Cherezov V, Stevens RC. 2010. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071.
- Xie H, Patching SG, Gallagher MP, Litherland GJ, Brough AR, Venter H, Yao SYM, Ng AML, Young JD, Herbert RB, Henderson PJF, Baldwin SA. 2004. Purification and properties of the Escherichia coli nucleoside transporter NupG, a paradigm for a major facilitator transporter sub-family. Mol Memb Biol 21:323–336.
- Yang J, Ma Y-Q, Page RC, Misra S, Plow EF, Qin J. 2009. Structure of an integrin αIIbβ3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci USA 106:17729–17734.
- Yildiz O, Vinothkumar KR, Goswami P, Kühlbrandt W. 2006. Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. EMBO J 25:3702–3713.
- Yu L, Sun C, Song D, Shen J, Xu N, Gunaskera A, Hajduk PJ, Olejniczak ET. 2005. Nuclear magnetic resonance structural studies of a potassium channel-charybdotoxin complex. Biochemistry 44:15834–15841.
- Zhou Y, Cierpicki T, Flores Jimenez RH, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. 2008. NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalysed disulphide bond formation. Mol Cell 31:896–908.