Abstract
Equilibrative Nucleoside Transporters (SLC29) are a family of proteins that transport nucleosides, nucleobases and nucleoside analogue drugs across cellular membranes. ENT1 is expressed ubiquitously in mammalian tissues and responsible for a significant portion of nucleoside analog drug uptake in humans. Despite the important clinical role of ENT1, many aspects of the regulation of this protein remain unknown. A major outstanding question in this field is the whether ENT1 is phosphorylated directly. To answer this question, we overexpressed tagged human (h) and mouse (m) ENT1, affinity purified protein using the tag, conducted phosphoamino acid analysis and found that m/hENT1 is predominantly phosphorylated at serine residues. The large intracellular loop of ENT1, between transmembrane domains 6 and 7, has been suggested to be a site of regulation by phosphorylation, therefore we generated His/Ubiquitin tagged peptides of this region and used them for in vitro kinase assays to identify target serines. Our data support a role for PKA and PKC in the phosphorylation of ENT1 within the intracellular loop and show that PKA can phosphorylate multiple sites within this loop while PKC specifically targets serines 279 and 286 and threonine 274. These data demonstrate, for the first time, that ENT1 is a phosphoprotein that can be directly phosphorylated at several sites by more than one kinase. The presence of multiple kinase targets within the loop suggests that ENT1 phosphorylation is considerably more complex than previously thought and thus ENT1 may be subject to phosphorylation by multiple pathways.
Keywords::
Introduction
The Equilibrative Nucleoside Transporters (ENTs) are integral membrane proteins which play a significant role in cellular physiology by facilitating the translocation of nucleosides across cellular membranes (King et al. Citation2006). To date, four isoforms of ENTs have been identified in mammals and the first isoform to be cloned, ENT1 (Griffiths et al. Citation1997), is the best characterized with regard to structure and function. ENT1 can be differentiated from other ENT family members based on its high affinity (nanomolar concentrations) for the inhibitor nitrobenzylthioinosine (NBTI). The ENTs are also important clinically since they transport nucleoside analogue drugs used in the treatment of viral infections and cancers (Galmarini et al. Citation2002, Young et al. Citation2008).
The putative two-dimensional topology of ENT1 predicts that the protein consists of 11 transmembrane domains, with a glycosylation site located within the first extracellular loop, an intracellular N-terminus and an extracellular C-terminus. The protein is predicted to have a large intracellular loop between transmembrane domains 6 and 7 (Robillard et al. Citation2008). Recent studies also suggest motifs, within the protein sequence, which aid in processes such as folding, trafficking and inhibitor sensitivity (Sundaram et al. Citation2001, SenGupta et al. Citation2002, SenGupta and Unadkat Citation2004, King et al. Citation2006, Lee et al. Citation2006, Nivillac et al. Citation2009). However, to date, our understanding of many aspects of ENT1 regulation, including the existence and physiological relevance of post-translational modifications such as phosphorylation, remains very limited.
During phosphorylation, a phosphate (PO4 -) group (usually donated by ATP) is added to a target protein (Seo and Lee Citation2004). It has been suggested that ∼ 30% of all eukaryotic proteins undergo phosphorylation at any given time within the cell (Gunawardena Citation2005). Specific kinases and phosphatases are responsible for the addition and removal of phosphate groups and the balance of phosphorylation/dephosphorylation can act as regulatory switches for a variety of proteins (Grimsrud et al. Citation2010). Phosphorylation most commonly occurs at serine residues but also at threonine residues and more rarely, tyrosine residues (Macek et al. Citation2007) via direct interaction of the kinase with the protein (López-Corcuera et al. Citation2001, Butt and Pitman Citation2005). Various studies have implicated phosphorylation as a regulatory mechanism for membrane proteins, such as Aquaporin 2 (AQP2), the Serotonin transporter (SERT) and the Dopamine transporter (DAT). For these membrane proteins, phosphorylation occurs predominantly at serine residues present in intracellular (cytoplasmic) portions of the protein (Procino et al. Citation2003, Jayanthi et al. Citation2005, Gorentla et al. Citation2009). The effects of phosphorylation can include conformational changes to the protein's structure which can influence a whole range of functions from protein-protein interactions to activity and trafficking (Procino et al. Citation2003, Mohapatra et al. Citation2007).
While previous data have suggested that kinase activity (e.g., by PKA, PKC and CKII) may have an influence on ENT1 surface expression and nucleoside transport (Sen et al. Citation1998, Coe et al. Citation2002, Bone et al. 2007), no information exists regarding the potential direct phosphorylation of ENT1. Investigations into the regulation of the ENT family are challenging due to the low endogenous expression levels within the cell and the hydrophobic nature of the protein.
To resolve the fundamental question of whether ENT1 is a phosphoprotein, we analyzed the phosphorylation status of transiently transfected heterologously expressed ENT1 and determined that constitutive phosphorylation occurs predominantly at serine residues in both mouse (m) and human (h) ENT1. Our data also suggest that a threonine residue is phosphorylated in mENT1 but possibly not hENT1, based on the presence of a weak signal for this residue for mENT1 following phosphoamino acid analysis. We also investigated the position of putative target sites for phosphorylation using in vitro kinase assays and site directed mutagenesis. We found that that the large intracellular loop connecting transmembrane domains 6 and 7 of mENT1 and hENT1 is phosphorylated by both PKC and PKA and that phosphorylation occurs at several sites.
Our demonstration that ENT1 is a phosphoprotein is an important breakthrough in our overall understanding of ENT1 regulation. The observation of multi-site phosphorylation was unexpected and suggests that regulation of ENT1 by this post-translational modification is more complex than previously suspected. However, since phosphorylation can be a key step in the regulation of many membrane proteins, identifying the target site(s) and kinase(s) involved in ENT1 phosphorylation will aid in our understanding of the mechanisms involved in the regulation of the ENT1 protein.
Materials and methods
Cell culture and transfection
COS-7 cells were grown in 10 cm2 plates in Dulbeccos Modified Eagle's Medium (DMEM, Gibco BRL) supplemented with 10% (v/v) fetal bovine serum (FBS, Wisent Inc., St Bruno QC, Canada) at 5% CO2 in a 37°C humidified incubator.
Prior to transfection, cells were grown to 80% confluence in 10 cm2 plates and were transfected with 3 × FLAG-m/hENT1. Transfections were carried out as follows: Lipofectamine2000 (10 μl, Invitrogen; Burlington, ON, Canada) was mixed with serum free DMEM (90 μl). In a separate vial, 10 μg of plasmid DNA was mixed with serum-free DMEM to a final volume of 100 μl. Both vials was incubated at room temperature for 5 min and then subsequently mixed and incubated at room temperature for 20 min. The media on each plate was replaced with 3 ml of serum free DMEM and the transfection mixture was added to the cells and allowed to sit at 37°C for 5 h. Serum-free DMEM was subsequently replaced with 10 ml DMEM supplemented with 10% (v/v) FBS and cells were left at 37°C for ∼ 20 h.
In vivo 32P-orthophosphate labeling of ENT1
Following transfection, the cells were gently rinsed with phosphate-free DMEM medium (pH 7.4, 2 ml × 3 cycles; Gibco) and incubated in fresh phosphate-free medium supplemented with 32P radioisotope-labeled orthophosphate for 7–8 h at 37°C (32PO4 -, 1 or 5 mCi/plate; 5mCi (185 MBq)/vial; Perkin Elmer, Waltham, MA, USA). Over-expression of non-labeled protein was conducted in a similar manner except that cells were incubated in medium lacking the radioisotope. Following incubation, cells were rinsed with ice-cold 1 × PBS (2 ml × 3 cycles) to remove excess radiolabel and collected by scraping in 1 × PBS followed by low-speed centrifugation (750 g). Pellets were stored at −80°C for less than 3 days before further analysis. Negative controls were included in all trials and consisted of the construct pEGFP-N, which encodes an amino-terminal GFP-tagged hENT1 protein under the same CMV promoter as pN-3 × FLAG-hENT1/mENT1, thus providing comparable transfection and over-expression capabilities. This construct was used to evaluate transfection efficiencies by fluorescence microscopy. Additionally, the over-expressed protein (GFP-tagged hENT1) was used as a negative control in anti-FLAG immuno-detection and autoradiography. As a positive control, the pCMV10-3 × FLAG-MEKK3 construct encoding a constitutively phosphorylated recombinant kinase protein, MEKK3, was used (Torres Citation2006). This construct was used as a positive control for FLAG purification and autoradiography.
Purification of 3 × FLAG tagged radiolabeled mENT1
FLAG purification of 32PO4 - labeled ENT1 was conducted primarily on solubilized membrane preparations although crude cell lysates were also used to ensure the cytoplasmic MEKK3 could be detected. Proteins were maintained at 4°C or below at all times to prevent protein degradation. To extract FLAG tagged protein, cell pellets were resuspended in an appropriate volume of 1 mM NaHCO3 buffer containing protease inhibitors and phosphatase inhibitors (200 μM NaVO4, 25 mM β-glycerol phosphate and 25 mM NaF – Sigma, St Louis, MO, USA). Cell lysis was accomplished using a 25 gauge needle (20 passes) and 5 min incubations in a sonication bath (three cycles of each). Following addition of an appropriate volume of 130 mM Na2CO3, each lysate was incubated at 4°C for 30 min. Lysates were centrifuged at 3000 g for 10 min, to pellet unlysed cell debris and the resulting supernatant was centrifuged at 60,000 g for 20 min. The resulting crude membrane pellet was solubilized overnight at 4°C in lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1.2% (w/v) Foscholine12, phosphatase and protease inhibitors).
The following day, solubilized membranes were added to anti-FLAG beads equilibrated at 4°C overnight in 1% (w/v) BSA in TBS (pH 7.4). Protein was allowed to bind to beads at 4°C overnight with rotation. After washing beads 6 times (10 min each) with lysis buffer, protein was eluted at 90°C for 10 min using standard 1 × protein loading buffer without DTT (to minimize IgG contamination). Samples were loaded onto a 7.5% (w/v) SDS gel (0.75 mm) and protein separation was accomplished using 200 V for 3.5 h. After SDS-PAGE the gel was stained with coomassie brilliant blue, dried and exposed to standard imaging film and a 32P intensifying screen (Kodak) at −80°C for ∼ 24 h.
Phosphoamino acid analysis
Following detection of 32PO4 - labeled proteins by autoradiography, protein bands were excised and subjected to phospho-amino acid analysis as previously described (Saxena and Kaur Citation2006). In all cases, signals greater than 200 counts/min (Geiger counts) were confirmed before proceeding to further analysis. Release of 32PO4 - labeled amino acids from the proteins trapped in the PVDF membrane fragments was carried out by acid hydrolysis in screw-cap Eppendorf tubes. Membrane fragments were submerged in 6 M HCl for 1 h at 110°C, followed by low speed-centrifugation (15,000 g, 10 s) to compact the PVDF along with any loose debris. The remaining hydrolysates were then dried overnight on a refrigerated (4°C) SpeedVac evaporator (Eppendorf; Mississauga, ON, USA). The dried samples were suspended in 10 μl of deionized H2O, mixed by gentle vortexing (30 min, 4°C) and centrifuged (15,000 g, 10 s) to remove debris. Aliquots (2–8 μl) of this solution were spotted on thin layer cellulose plates followed by layering of a 1–2 μl mixture of non-labeled phospho-amino acid standards (1 mg/ml each of P-Ser, P-Thr and P-Tyr (Sigma) dissolved in HTLE Buffer pH 1.9 (50 ml formic acid (88% w/v), 156 ml glacial acetic acid, 1794 ml deionized water). Separation of phospho-amino acids, 32PO4 - labeled and standards was conducted in two dimensions using a Hunter Thin-Layer cellulose electrophoresis (HTLE) unit (CBS Scientific; Del Mar, CA, USA). Segregation of amino acids was first conducted in HTLE Buffer pH 1.9 at 1.5 kV for 35 min followed by a second perpendicular displacement in HTLE Buffer pH 3.5 (100 ml glacial acetic acid, 10 ml pyridine, 1890 ml deionized water) at 1.3 kV for 20 min. After allowing the plates to fully dry, amino acid standards were visualized by staining with ninhydrin (0.25% (v/v) in EtOH; Sigma) and 32PO4 - labeled amino acids were detected with autoradiography as described above with incubations of 14 days at −80°C.
Predicting phosphorylation sites in 3 × FLAG-ENT1
Having determined the residue involved in the phosphorylation of ENT1, sequence alignments of 3 × FLAG-mENT1 and 3 × FLAG hENT1 were made using ClustalW (European Bioinformatics Institute – http://www.ebi.ac.uk/clustalw). The resulting output was then analyzed to identify the intracellular serines which are conserved in human and murine ENT1.
The same sequences were analyzed using NetPhosK 1.0 (Centre for Biological Sequence Analysis, Technical University of Denmark DTU – http://www.cbs.dtu.dk/services/NetPhosK/). This program predicts the potential phosphorylation sites within a protein along with the putative kinases responsible for facilitating this phosphorylation. This allowed us to predict which of the serines identified above where likely to be kinase targets and which kinases to test.
Synthesis of ENT1 intracellular loop peptides
Given that previously published literature implicated the large intracellular loop as being involved in kinase mediated regulation of ENT1, plasmids (pJExpress 401 under T5 promoter control) containing the intracellular loop of hENT1 (residues 228–290) and mENT1 (residues 228–287), preceded by His and Ubiquitin tags, were synthesized by DNA 2.0 (Menlo Park, CA, USA) resulting in the following peptides:
hENT1: HHHHHHMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGGLVPRGSRLEFYRYYQQLKLEGPGEQETKLDLISKGEEPRAGKEESGVSVSNSQPTNESHSIKAILKNIS
mENT1: HHHHHHMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGGLVPRGSRTEFYRHYLQLNLAGPAEQETKLDLIREEPKGRREESGVPGPNSPPTNRNQSIKAILKSI
The portion of the peptide corresponding to the ENT1 intracellular loop is indicated in bold. Plasmids were transformed into Escherichia coli BL21 bacteria followed by plating onto kanamycin (50 μg/ml) plates. Following overnight incubation at 37°C, a single colony was inoculated into 3 ml LB containing 50 μg/ml of kanamycin and grown at 37°C for 8 h. The resulting starter culture was subsequently transferred to 50 ml LB containing 50 μg/ml kanamycin and allowed to grow for a further 14 h before being transferred to 1 l of M9 minimal medium [0.5 M D-glucose, 0.02 M Na2HPO4, 0.02 M KH2PO4, 0.01 M NaCl, 0.02 M NH4Cl supplemented with 0.1 mM CaCl2, 1 mM MgSO4, 50 μg/ml kanamycin and trace metals as follows: 3.07 mM FeCl3, 0.37 mM ZnCl2, 0.06 mM CuCl2, 0.04 mM CoCl2, 0.16 mM H3BO3, 6.82 mM MnCl2]. When the bacterial culture reached an OD600 = 0.7, 1 mM IPTG was added to induce protein expression. Bacteria were induced at 37°C for 2 h and then pelleted at 7,000 g for 20 min. Pellets were resuspended in 30 ml T300 [10 mM Tris-HCL, 0.5 M NaCl; pH 8] and passed through a French press (5 times) to lyse bacteria and free protein. Unbroken cell debris was pelleted at 16,000 g for 20 min.
His-Ubiquitin tagged peptides were extracted from the supernatant (obtained from the previous centrifugation step) using standard nickel affinity chromatography methods. Briefly, following preparation of the column, protein was bound at a rate of 2 ml/min. The column was then washed at a rate of 5 ml/min first with 100 ml T300 and then with 100 ml T300 buffer supplemented with 10 mM imidazole. Bound protein was eluted from the column using 50 ml T300 buffer supplemented with 20 mM EDTA. Presence of the peptide was confirmed using a 10% (w/v) SDS gel and Coomassie blue staining.
The 50 ml sample was then concentrated at 4000 g to 4 ml using Microcon YM-30 columns (Millipore) and subjected to further purification by fast protein liquid chromatography (FPLC) using a Sephadex S-100 16/60 column (GE Biosciences). The resulting elution fraction was concentrated to 5 ml using Microcon YM-30 columns.
In vitro phosphorylation of the ENT1 intracellular loop peptide
In vitro phosphorylation assays were conducted using 10 μg of the ENT1-loop peptide, which was treated with PKC (obtained from rat brain; 0.08 units) or PKA (catalytic subunit from mouse; 5000 units) (Calbiochem, San Diego, CA, USA) and [γ32P]-ATP (5 mCi/ml; for autoradiography) or non-radiolabeled ATP (for mass spectrometry). Following 2 h incubation at 30°C, samples were mixed with an appropriate volume of 6 × PLB and boiled at 90°C for 10 min. Protein samples were separated by 10% SDS-PAGE for 3.5 h at 200 V.
Following separation, 16 kDa bands representing phosphorylated or non-phosphorylated mENT1 and hENT1 peptides were visualized using Coomassie blue stain. Gels containing radiolabeled protein were dried and subjected to autoradiography using standard imaging film in cassettes lined with a 32P intensifying screen for 1 h at room temperature or overnight at −80°C. Gels containing non-radiolabeled samples were prepared for mass spectrometry analysis.
In vitro phosphorylation of mutated mENT1 loop peptides
Having determined that the ENT1 large intracellular loop is indeed phosphorylated, we then determined which specific residues are involved. Following analysis of the ENT1 sequence and based on in silico predictions, four serines (S264, S271, S279 and S286) and one threonine (T274) (in the mENT1 sequence) were suggested as putative targets for phosphorylation within the large intracellular loop. Therefore, in order to determine which residue(s) serve as targets of phosphorylation, each serine or the threonine was mutated to create mENT1 peptides containing serine (or threonine) to alanine amino acid substitutions. All mutations were introduced using the Stratagene Quikchange site directed mutagenesis kit (Stratagene, CA, USA) into the His- and ubiquitin-tagged mENT1 loop template using the primers indicated in and following the manufacturer's instructions.
Table I. List of primers used to design the mENT1 serine to alanine subsitution mutants. All primers are shown in the 5′–3′ direction.
Positive clones, obtained following transformation and plating on LB-agar containing 50 μg/ml ampicillin, were sequenced in both forward and reverse directions at the York University Core Molecular Sequencing Facility. Samples showing the correct mENT1 sequence (serine to alanine substitutions only and no other mutations) were purified using the Endo-Free Maxi prep Kit (Qiagen Inc., Canada).
In-gel trypsinization and mass spectrometry
Protein bands of the expected size for the ubiquitin tagged intracellular loop peptide (∼16 kDa) were excised from the Coomassie blue stained gels and diced into small fragments (∼ 1 mm3). Coomassie blue stain was removed by boiling gel fragments at 60°C for 20 min in the presence of 30% (v/v) ethanol (three cycles until stain was completely removed).
Destained gel pieces were washed once with 50 mM ammonium bicarbonate and dehydrated with 100% (v/v) acetonitrile. Dehydrated gel fragments were then dried completely by vacuum centrifugation. Digestion of peptide within the gel fragments was achieved using 1 μg of porcine trypsin (Promega) dissolved in 50 mM ammonium bicarbonate (4 h at 37°C). The resulting eluates, containing tryptic peptides, were transferred to a separate tube and gel fragments were incubated at room temperature with 25 mM ammonium bicarbonate (20 min). The supernatant was added to the tryptic peptide eluate. Finally, gel fragments were incubated with 5% (v/v) formic acid with 50% (v/v) acetonitrile (3 × 20 min). The supernatant was added to the existing peptide eluate each time. The eluate was then dried completely by vacuum centrifugation. In preparation for desalting, the dried sample was resuspended in 20 μl 0.1% (v/v) trifluoroacetic acid (TFA).
For desalting, C18 ZipTips were used according to manufacturer's instructions. Elution was accomplished using 3 μl of matrix solution containing α-cyano-4-hydroxycinnamic acid in 65% (v/v) acetonitrile and 0.3% (v/v) TFA. The eluate was spotted onto a sample plate and analyzed using MALDI-TOF mass spectrometry on a QSTAR XL instrument (AB MDS/SCIEX, Vaughan, ON, Canada). Peaks of interest were further analyzed by MALDI-MS/MS and the resulting spectra were analyzed using Mascot (Matrix Science – www.matrixscience.com) and Protein Prospector software (University of California San Francisco Mass Spectrometry Facility – www.proteinprospector.ucsf.edu).
Results
In vivo phosphorylation of ENT1
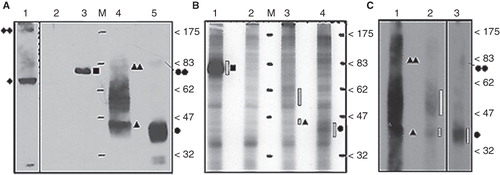
32PO4 - labeled FLAG tagged recombinant proteins were initially isolated from crude cell lysates using the FLAG purification system and detected with autoradiography (, Panel B). Signals produced by 32PO4 - labeled recombinant protein for N-3 × FLAG-MEKK3, -hENT1 and -mENT1 purifications correspond to protein profiles on immuno-blots (, Panel A). The autoradiographic data confirmed that the positive control N-3 × FLAG-MEKK3 is constitutively phosphorylated (as predicted) and suggests that N-3 × FLAG-hENT1 and N-3 × FLAG mENT1, when transfected into cultured cells, can be directly phosphorylated. However, to ensure that the ENT1 results were not due to band contamination as a result of inefficient FLAG purification, the procedure was repeated with membrane preparations (rather than total cell lysates) made from larger volumes of transfected cells and additional wash steps were incorporated into the FLAG purification. These modifications significantly enhanced the signal and confirmed the presence of purified phosphorylated ENT1 proteins (, Panel C). Autoradiographic signals were confirmed by matching immunoblots loaded with unlabelled samples (, Panel A). Signals that we suspect represent ENT1 dimers or protein complexes (∼ 83 kDa) and hENT1 aggregation products (47–62 kDa) were identical in both immunoblots and autoradiographs.
Figure 1. In vivo 32PO4- labeled N-3 × FLAG-hENT1 and mENT1 extracted from crude lysates and total-membrane enriched lysates. (A) Anti-3 × FLAG immuno-detection of tagged recombinant proteins from crude lysates prepared from non-labeled cells (1.2 μg total protein per lane). Lane 1: GFP-hENT1 (diamond) detected with anti-GFP antibodies. Lane 2: GFP-hENT1 (negative control). Lane 3: N-3 × FLAGMEKK3 (positive control, square). Lane 4: N-3 × FLAGhENT1 (triangle). Lane 5: N-3 × FLAG-mENT1 (circle). Putative dimers or protein complexes of ENT1s (double symbols) are present along with hENT1 aggregated/variably glycosylated products (between the 47–83 kDa markers). (B) Autoradiograph of FLAG-purified 32PO4-labeled recombinant proteins from crude cell lysates. Lane 1: 32PO4- labeled N-3 × FLAGMEKK3 (positive control). Lane 2: GFP-hENT1 (negative control). Lane 3: 32PO4- labeled N-3 × FLAG-hENT1. Lane 4: 32PO4- labeled N-3 × FLAG-mENT1. Presence of 32PO4- labeled recombinant protein is emphasized with dashed bars. Each lane was loaded with equal amounts of purified protein. (C) Autoradiography establishes the presence of 32PO4- labeledN-3 × FLAG-hENT1 and mENT1, purified from total membrane enriched lysates. 32PO4- labeled protein is indicated by dashed bars. Lanes 1 and 2: N-3 × FLAG-hENT1, ∼ 10 μg and 2.5μg. Lane 2: Loaded with less labeled protein to avoid interference by film overexposure. Monomeric and putative dimer complexes are observed as broad bands indicated by filled single and double symbols, while aggregated/ variably glycosylated products are present as dark smears. Lane 3: N-3 × FLAG-mENT1, ∼ 10 μg total protein. Representative images are shown based on five independent experiments with similar results.
Phosphoamino acid mapping
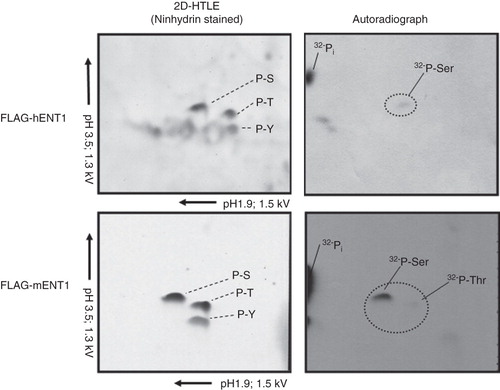
Having confirmed that N-3 × FLAG-hENT1/mENT1 can be labeled metabolically with 32PO4 -, phosphoamino acid analysis was conducted to determine the target residue. Autoradiographic data show that N-3 × FLAG-hENT1 and mENT1 are directly phosphorylated on serine residues (). These results were highly reproducible and the target amino acid was confirmed by comparison with ninhydrin stained standards (). A faint signal for a radio-labeled threonine was also detected for mENT1 but it was not as reproducible as the serine signal.
Figure 2. Phosphoamino acid analysis using of 32PO4 - labeled 3 × FLAG-mENT1 and 3 × FLAG-hENT1. Hunter Thin Layer Electrophoresis of 3 × FLAG-mENT1 and 3 × FLAG-hENT1. Samples stained with ninhydrin show the location of non-radiolabeled phosphoamino acid standards (Serine – S, Threonine – T or Tyrosine – Y). Autoradiographs show the position of 32PO4 - labeled amino acids. The phosphorylated residues were determined by comparing the autoradiograph to the ninhydrin stained standards and these data suggest that phosphorylation occurs on serines for both 3 × FLAG-hENT1 and 3 × FLAG-mENT1. Additionally, a weak signal is seen for threonine for 3 × FLAG-mENT1. Labeled inorganic phosphate (32Pi), a sample processing degradation product, was also observed. Representative images are shown based on two independent experiments.
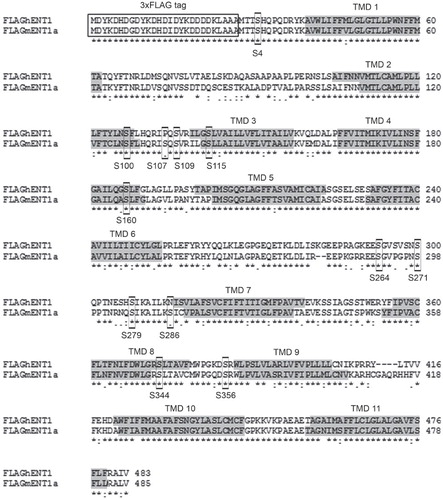
Despite having identified the phosphorylated residue, the location of the phosphorylated serine(s) (and possibly threonine) was unknown. Phosphorylation sites on other membrane transport proteins (e.g., serotonin and dopamine transporters), are present in intracellular regions such as the N- or C-termini or within loop regions connecting the transmembrane domains (Khoshbouei et al. Citation2004, Jayanthi et al. Citation2005). Since ENT1 possesses a short intracellular N-terminal region and an extracellular C-terminus, we hypothesized that the most likely target site for phosphorylation was the large intracellular loop between their 6th and 7th transmembrane domains, which has previously been implicated in ENT1 regulation (Stolk et al. Citation2005, Robillard et al. Citation2008). In silico analyses of the entire sequence of hENT1 and mENT1 indicated that there were 12 intracellular serines, which were putative kinase targets () including a number in the intracellular loop.
Figure 3. ClustalW alignment of 3 × FLAG-hENT1 and 3 × FLAG-mENT1. The 3 × FLAG-tag is indicated with a box while the transmembrane domains are indicated in gray. The 12 predicted serine targets are indicated with boxes. High identity of residues is indicated as (*) while high homology is indicated as (:).
In vitro kinase assays of the ENT1 large intracellular loop
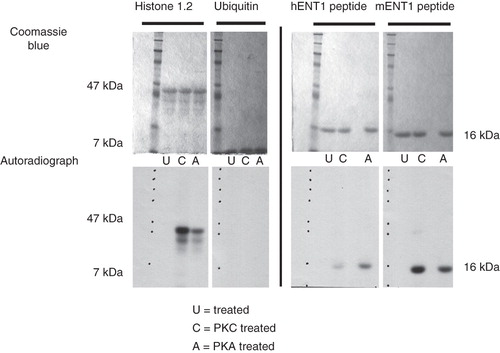
Previous reports suggest that PKC, PKA and CKII may be involved in the regulation of ENT1 (Coe et al. Citation1996, Citation2002, Bone et al. 2007). However, we tested only PKA and PKC since we were working with the mENT1a, which does not contain a previously identified CKII consensus site (Bone et al. 2007). Our data demonstrate that phosphorylation of the intracellular loop in the presence of PKC or PKA for both mENT1 and hENT1 (). This phosphorylation was not occurring within the ubiquitin tag, since no signal was detected in samples containing the ubiquitin tag alone. These data confirm that there are residues within the intracellular loop of ENT1, which can be phosphorylated by PKC or PKA.
Figure 4. In vitro kinase assays of the Ub-mENT1 and Ub-hENT1 large intracellular loop peptides using PKA or PKC. Peptides of the large intracellular loops were tested for phosphorylation activity using PKA (A) or PKC (C) and as a negative control, an untreated sample (U). Coomassie blue stained gels confirm equal loading. The soluble protein, histone 1.2, was used a positive control and shows phosphorylation by PKC and PKA. To determine whether the kinases were able to label the ubiquitin tag at the N-terminal end of each peptide, the ubiquitin backbone alone was tested for phosphorylation activity. No signals are seen demonstrating that the ubiquitin backbone does not contribute to phosphorylation of the peptide. Autoradiography of the hENT1 and mENT1 peptides indicates that both are phosphorylated by PKA and PKC. Assays were conducted twice with identical results. Representative images are shown based on two independent experiments.
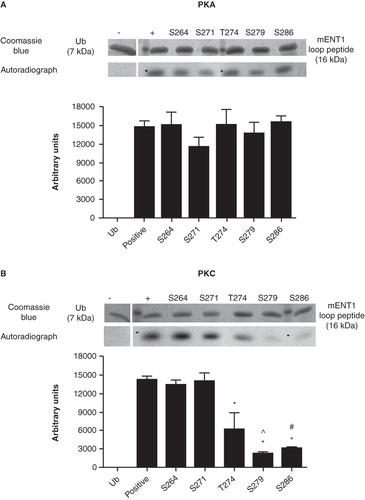
To identify specific target residue(s), we conducted in vitro kinase assays with PKA on mutated mENT1 loop peptides each containing a single alanine substitution at one of the four serines (S264, S271, S279 and S286) or threonine (T274) within this region. We found that none of the single substitution mutants resulted in a decrease or loss of phosphorylation (). These data suggest that phosphorylation of mENT1 by PKA cannot be assigned to a single serine residue but rather that multiple residues can be phosphorylated by this kinase. Furthermore, the phosphorylation seen in the control was not occurring on the single threonine residue since mutation of T274 did not result in the decrease or complete loss of phosphorylation by PKA, suggesting that phosphorylation by this kinase occurs at other sites. However, mutation of serines 279 and 286, and, to a lesser extent, threonine 274, did result in a significant decrease in phosphorylation by PKC suggesting that these are targeted by this kinase. In contrast, no change in the level of phosphorylation was seen, compared to control, when serines 264 and 271 were mutated () suggesting that these are not targets for PKC under these conditions.
Figure 5. In vitro PKA and PKC kinase assays of single residue substitution mutants of the large intracellular loop peptide. Single mutations were introduced into the mENT1 loop peptide to substitute each of the four predicted serine residues and the one predicted threonine residue with an alanine. All mutants were subsequently subjected to an in vitro kinase assay using PKA or PKC to determine whether the mutation affects phosphorylation. The ubiquitin back bone was used as a negative control. (A) Autoradiography and densitometric analysis show that no single mutation affects the ability of PKA to phosphorylate mENT1 compared to control. (B) Autoradiography and densitometric analysis show that mutations in serines 264 and 271 do not affect phosphorylation of mENT1, by PKC, when compared to control. However, mutation of threonine 274, serine 279 or serine 286 does result in a significant decrease in phosphorylation compared to control. (*) p < 0.001 versus positive control, (^) p < 0.001 vs. T274 and (#) p < 0.01 vs. T274. Values are based on three independent experiments and are represented as mean ± SEM.
To determine the likelihood of specific residues being targeted by specific kinases, we conducted in silico predictions of the ‘strength’ of consensus sites in the intracellular loop using NetPhosK 1.0. The resulting output assigns the highest scores, for potential phosphorylation by PKA, to serines 264, 271 and 286 (). Interestingly, NetPhosK 1.0 predictions for PKC consensus strength are consistent with our in vitro data in that serine 279 and threonine 274 (to lesser extent) have the highest confidence scores (). Intriguingly, serine 286 is not predicted as a strong candidate for phosphorylation by PKC by NetPhosK 1.0. Taken together, our data show that phosphorylation of mENT1 by PKA and/or PKC occurs at multiple sites.
Table II. NetPhosK 1.0 prediction results for the mENT1 loop peptide. Predictions of putative phosphorylation sites within the mENT1 large intracellular loop for both PKA and PKC. Confidence scores are listed for each residue with the highest possible confidence equal to 1.0 and the lowest equal to 0.0. Residues at putative consensus sites scoring above the arbitrary confidence limit of 0.5 are shown in bold. Residues that have been shown to have an importance in phosphorylation in vitr o are indicated with a star.
Mass Spectrometric Analysis of the hENT1 and mENT1 intracellular loop peptides
Following identification of the putative target sites for phosphorylation we subjected the control peptide to in-gel trypsinization assays combined with MALDI-TOF mass spectrometry in order to determine sequences which may contain phosphorylated residues. This approach proved to be unsuccessful for analysis of the hENT1 peptide since we were unable to obtain any coverage of the hENT1 protein itself, but rather only of the ubiquitin backbone. However, mass spectrometric analysis of the mENT1 peptide resulted in coverage of almost the entire peptide with exception of the last 11 residues.
Analysis of MALDI-TOF spectra obtained from the control mENT1 peptide, incubated with PKA, reveal the presence of a 1693.8 m/z peak, which corresponds to the sequence REESGVPGPNSPPTNR (). This peptide sequence contains Serines 264 and 271 as well as Threonine 274. Analysis of this peak (using MS/MS) confirmed it as mENT1. During phosphorylation, each phosphate group that is added to the target protein will result in an 80 dalton (Da) increase from its original mass. Further analysis of the spectrum reveals that a second peak of 1773.8 m/z appears. Since the shift from 1693.8–1773.8 is only 80 Da, this would suggest that only a single residue is being phosphorylated in this region (either at S264, S271 or T274) ( inset). The intensity of the 1773.8 peak is significantly less than the 1693.8 unphosphorylated peak and we were, therefore, unable to identify it using MASCOT software following MS/MS analysis. However, when the 1773.8 peak was subjected to further fragmentation during MS/MS, the resulting spectrum contained the 1693.8 peak, which would imply a loss of the phosphate group (). The observation of the shift from the 1693.8 to the 1773.8 peak shows that this approach is feasible in elucidating residues involved in PKA-dependent phosphorylation. However, due to a lack of coverage of the last 11 amino acids, we cannot rule out that S279 or S286 may also be targeted by PKA and this would not be unexpected given our previous findings with the kinase assays, which suggest that mENT1 is phosphorylated by PKA at multiple sites.
Figure 6. Mass spectrometry analysis of the mENT1 intracellular loop peptide treated with PKA. A representative image is shown of the spectrum obtained following tryptic digest of PKA treated mENT1 loop peptide. The 1693.8 peak is indicated with a black arrow and represents the sequence REESGVPGPNSPPTNR. MASCOT MS/MS analysis of this peak resulted in a high score value for this sequence and matched it to endogenous mENT1. The second peak obtained in this spectrum was 1773.8 which results from the 80 dalton shift following addition of a single phosphate group. The 1773.8 peak is significantly lower in intensity compared to the 1693.8 peak due to the high intensity of the 1693.8 peak however this peak is clearly present and above background when magnified (see inset).
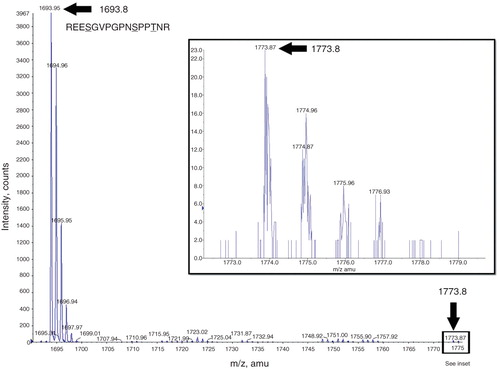
Figure 7. MS/MS analysis of the 1773.8 m/z peak. A representative image of MS/MS analysis of the 1773.8 m/z peak obtained from tryptic digest of the mENT1 peptide following PKA treatment. In this spectrum, both the 1773.8 and the 1693.8 peak are visible which would suggest both the phosphorylated and unphosphorylated forms of the REESGVPGPNSPPTNR fragment. Despite obtaining both peaks, MASCOT MS/MS analysis was unable to match the 1773.8 peak to the mENT1 sequence.
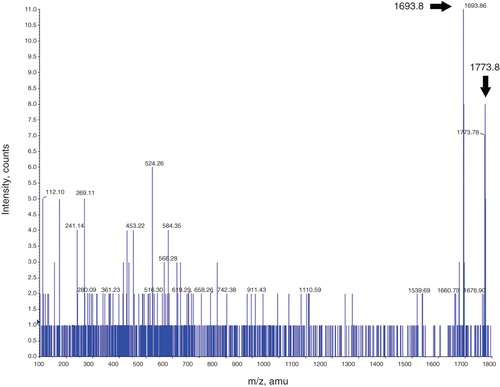
Mass spectrometric analyses of the control mENT1 peptide, with PKC, show the same spectra as those treated with PKA. In samples treated with PKC we were still unable to obtain coverage of the last two serine residues. Therefore, we cannot make any statements regarding their correlation with the radiolabeled kinase assays.
Discussion
There are currently no reports in the literature demonstrating that ENT1 is phosphorylated and, therefore, no information exists on the residues targeted by phosphorylation, which may be involved in ENT1 regulation. While various studies have implicated a number of different kinases as regulators of the ENT1 protein (Coe et al. Citation1996, Citation2002, Stolk et al. Citation2005, Bone et al. 2007), there has been no evidence for a link between these kinases and direct phosphorylation of the ENT1 protein. Here, for the first time, we demonstrate that heterologously expressed ENT1 is phosphorylated in vivo, and provide information on target residues and kinases that can phosphorylate the large intracellular loop which was identified as a putative site for kinase regulation when ENT1 was initially described (Griffiths et al. Citation1997). Our findings support the concept of the large intracellular loop as a location of direct phosphorylation based on in silico and in vitro data.
Although understanding the regulation of hENT1 is of specific clinical interest since it is a drug transporter, we have routinely found that mENT1 is more soluble and amenable to biochemical analysis by a variety of assays. Therefore, we used mENT1 to show that the protein is predominantly and directly phosphorylated within the large intracellular loop connecting transmembrane domains 6 and 7. These data support earlier studies on the mENT1 protein, which have implicated this region as being involved in regulation of mENT1 (Stolk et al. Citation2005, Bone et al. 2007) although these data reflect regulation of the mENT1b isoform which possesses a predicted CKII consensus sites, which is absent in mENT1a, used in our assays and we did not test CKII in our assays. The role of CKII remains to be determined but should be relatively straightforward using the approach described here.
Our data suggest that ENT1 can be phosphorylated by PKA or PKC. Differences in the ability of PKA and PKC to phosphorylate a target protein are due, in part, to differences in their consensus sites (PKA = R(R/K)Φ(S/T) or RRX(S/T) Φ – where Φ is a hydrophobic residue); (PKC = RXX(S/T)XRX – where X is any amino acid). However, when dealing with the potential for multisite phosphorylation one of the main factors that must be kept in mind is the number of sites which are involved and in what order they get phosphorylated. This is because the number of phosphorylation sites and their surrounding consensus sequences are defining factors for the order in which kinases and phosphatases will act upon the protein to induce a biological effect (Salazar and Höfer Citation2009).
The fact that we were not able to obtain the identity of the target sites for kinases by means of mass spectrometry could be due to the low ionization ability of phosphopeptides. This requires further optimization, perhaps with different matrices, to try and improve the ionization of phosphopeptides (Hou et al. Citation2010). However, our data suggest that multisite phosphorylation, by PKA, could involve serines 264 or 271 or threonine 274 in addition to serine 279 and/or 286. Further mutational analyses would be needed to determine which combinations of residues are contributing the PKA-mediated regulation.
We also show here that PKC phosphorylates three target residues S279, S286 and T274. These data raise the possibility of cooperative phosphorylation as a regulatory mechanism for ENT1 as has been demonstrated for the sodium-hydrogen exchanger regulatory factor-1 (NHERF-1), which exhibits hierarchical phosphorylation such that phosphorylation of a threonine residue (Thr95) leads to the phosphorylation of a serine residue (Ser77) and there is a functional consequence in terms of regulation of renal phosphate transport (Weinman et al. Citation2010). Multisite cooperative phosphorylation was also seen in studies on the serotonin transporter (SERT) where phosphorylation of a serine residue on SERT leads to a reduction in transport activity while subsequent phosphorylation of a threonine residue leads to enhanced endocytic activity while phosphatase activity reverses the endocytic effects (Jayanthi et al. Citation2005). Similar observations were reported for the human norepinephrine transporter (hNET) (Jayanthi et al. Citation2006). These findings suggest that regulation of a phosphoprotein is mediated by a balance between kinases and phosphatases at multiple sites within the protein's sequence as may be the case with ENT1.
Phosphorylation at a single site can lead to the adoption of a flexible or unfolded structure by the target protein so that several residues become equally accessible to the kinase (Mittag et al. Citation2010). The cystic fibrosis transmembrane conductance regulator (CFTR) comprises two transmembrane domains, two nucleotide binding domains and a regulatory domain which contains 10 consensus sites for phosphorylation. PKA phosphorylation of CFTR's regulatory domain resulted in a conformational change that caused this domain to be displaced or that conformational changes occur in nucleotide binding domains and transmembrane segments adjacent to the phosphorylation sites (Grimard et al. Citation2004). These structural changes were suggested to improve the ability of solutes to interact with the nucleotide binding domains (Grimard et al. Citation2004). Similar findings of regulation of substrate affinity through phosphorylation-induced conformational changes were also observed for the glutamate receptors and the organic cation transporters (Ciarimboli and Schlatter Citation2005, Wang et al. Citation2006).
Recent nuclear magnetic resonance (NMR) studies conducted by our laboratory have identified the large ENT1 intracellular loop as being unstructured and as having a flexible conformation (Reyes et al. Citation2011), which would promote recognition by kinases thereby facilitating rearrangements in a proteins conformation (Mittag et al. Citation2010). Phosphorylation of membrane proteins has also been shown to facilitate endocytosis as demonstrated by the dopamine transporter (DAT), which is phosphorylated on serines by PKC (Foster et al. Citation2002, Cervinski et al. Citation2010). Inhibition of phosphatases led to a decrease in the amount of DAT at the cell surface followed by intracellular sequestration and trafficking through the recycling and degradation pathways. The decrease in cell surface DAT was proposed to regulate dopamine clearance from the synaptic cleft (Foster et al. Citation2002, Cervinski et al. Citation2010). Aquaporin is phosphorylated on a serine residue, which results in the translocation to, and fusion with, the apical plasma membrane (van Balkom et al. Citation2002).
The consequences of direct phosphorylation of ENT1 remain unclear since a three-dimensional structure is not available and the mechanism of translocation of substrate remains unknown. Direct phosphorylation of ENT1 may regulate trafficking, interaction with other proteins, substrate translocation and/or affinity, inhibitor sensitivity and turnover at the membrane. Coordinated regulation of the sensitivity of the es transporter (now known to be ENT1) to a pharmacological inhibitor (ethanol) by PKC, PKA and PP1/2A was proposed prior to the cloning of ENT1 (Coe et al. Citation1996). Sensitivity of es- (ENT1-) dependent adenosine uptake to inhibition by ethanol requires activation of PKA while activation of PKC leads results in ethanol insensitive transport perhaps as a result of a subtle conformational change in the protein which affects the ethanol-interaction site on the protein. Phosphatase 1 or 2A activity, which is regulated by PKC, blocks the effects of PKA on ethanol sensitivity. Thus, the combination of kinase and phosphatase activities alters the response of the es transporter to ethanol and thus modulates adenosine reuptake leading to altered purinergic signaling through adenosine receptors and subsequent clinically relevant cellular responses to ethanol (Coe et al. Citation1996). It has also been proposed that PKC regulates pools of ENT1 transporters at the membrane and can modulate overall uptake by activating ‘inactive’ proteins without trafficking of subcellular pools (Coe et al. Citation2002, Fernández Calotti et al. Citation2008). These combined studies have been conducted in very different cell types, but taken together suggest that PKC may play an important role in the regulation of ENT1. Our own studies suggest that mutation of the residues identified as phosphorylation targets in this study results in equivocal changes in overall transport but a significant increase in NBTI binding (Coe et al., unpublished observations) suggesting that phosphorylation at one or more of these residues regulates access to a binding site or possibly transporter turnover (as previously described for PKC regulated endocytosis of DAT). Based on the data presented here, we can now speculate that direct phosphorylation of ENT1 by PKC may be a contributing mechanism to previously reported observations.
The observation that ENT1 is phosphorylated at a number of sites was unexpected and suggests that the regulation of ENT1 by phosphorylation is more complicated than we had previously anticipated. Correlating phosphorylation site and status of ENT1 with signaling cascades (such as adenosine receptor activation) and with changes in functional outcomes, such as rate of uptake, affinity, trafficking and recycling, will be a challenging but important area of research in the future.
Conclusion
ENT1 can be phosphorylated in vitro, at multiple sites and the large intracellular loop can be phosphorylated directly by both PKA and PKC at a number of serine residues. These data demonstrate that ENT1 is a phosphoprotein and suggest that it is subject to regulation directly by a number of kinases at a number of sites. These findings lay a foundation for a variety of studies that will link signaling pathways and regulation of function of ENT1.
Acknowledgements
We thank Dr Michael Scheid (York University, Canada) for providing the N-3 × FLAG-MEKK3 construct.
Declaration of interest: This research was supported by a Discovery Grant (RGPIN – 203397) to I.R. Coe from the Natural Sciences and Engineering Research Council, Canada. Infrastructure support to K.W.M. Siu was provided by the Canada Foundation for Innovation, the Ontario Innovation Trust, the Ontario Research and Development Challenge Fund, Applied Biosystems, MDS SCIEX, and Genome, Canada. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Butt SJ, Pitman RM. 2005. Indirect phosphorylation-dependent modulation of postsynaptic nicotinic acetylcholine responses by 5-hydroxytryptamine. Eur J Neurosci 21:1181–1188.
- Cervinski MA, Foster JD, Vaughan RA. 2010. Syntaxin 1A regulates dopamine transporter activity, phosphorylation and surface expression. Neuroscience 170:408–416.
- Ciarimboli G, Schlatter E. 2005. Regulation of organic cation transport. Pflugers Arch 449:423–441.
- Coe IR, Yao L, Diamond I, Gordon AS. 1996. The role of protein kinase C in cellular tolerance to ethanol. J Biol Chem 271:29468–29472.
- Coe I, Zhang Y, McKenzie T, Naydenova Z. 2002. PKC regulation of the human equilibrative nucleoside transporter, hENT1. FEBS Lett 517:201–205.
- Fernández Calotti P, Galmarini CM, Cañones C, Gamberale R, Saénz D, Avalos JS, Chianelli M, Rosenstein R, Giordano M. 2008. Modulation of the human equilibrative nucleoside transporter1 (hENT1) activity by IL-4 and PMA in B cells from chronic lymphocytic leukemia. Biochem Pharmacol 75:857–865.
- Foster JD, Pananusorn B, Vaughan RA. 2002. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem 277:25178–25186.
- Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR, Dumontet C. 2002. Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int J Cancer 97:439–445.
- Gorentla BK, Moritz AE, Foster JD, Vaughan RA. 2009. Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry 48:1067–1076.
- Griffiths M, Yao SY, Abidi F, Phillips SE, Cass CE, Young JD, Baldwin SA. 1997. Molecular cloning and characterization of a nitrobenzylthioinosine-insensitive (ei) equilibrative nucleoside transporter from human placenta. Biochem J 15:739–743.
- Grimard V, Li C, Ramjeesingh M, Bear CE, Goormaghtigh E, Ruysschaert JM. 2004. Phosphorylation-induced conformational changes of cystic fibrosis transmembrane conductance regulator monitored by attenuated total reflection-Fourier transform IR spectroscopy and fluorescence spectroscopy. J Biol Chem 279:5528–5536.
- Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, Coon JJ. 2010. Phosphoproteomics for the masses. ACS Chem Biol 5:105–119.
- Gunawardena J. 2005. Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc Natl Acad Sci USA 102:14617–14622.
- Hou J, Xie Z, Xue P, Cui Z, Chen X, Li J, Cai T, Wu P, Yang F. 2010. Enhanced MALDI-TOF MS analysis of phosphopeptides using an optimized DHAP/DAHC matrix. J Biomed Biotechnol 2010:759690.
- Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S. 2005. Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol 67:2077–2087.
- Jayanthi LD, Annamalai B, Samuvel DJ, Gether U, Ramamoorthy S. 2006. Phosphorylation of the norepinephrine transporter at threonine 258 and serine 259 is linked to protein kinase C-mediated transporter internalization. J Biol Chem 281:23326–23340.
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. 2004. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol 2:E78.
- King AE, Ackley MA, Cass CE, Young JD, Baldwin SA. 2006. Nucleoside transporters: From scavengers to novel therapeutic targets. Trends Pharmacol Sci 27:416–425.
- Lee EW, Lai Y, Zhang H, Unadkat JD. 2006. Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): Implications for interspecies differences in mitochondrial toxicity of fialuridine. J Biol Chem 281:6700–6706.
- López-Corcuera B, Aragón C, Geerlings A. 2001. Regulation of glycine transporters. Biochem Soc Trans 29:742–745.
- Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, Jensen PR, Mann M. 2007. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics 6:697–707.
- Mittag T, Kay LE, Forman-Kay JD. 2010. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit 23:105–116.
- Mohapatra DP, Park KS, Trimmer JS. 2007. Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation. Biochem Soc Trans 35:1064–1068.
- Nivillac NM, Wasal K, Villani DF, Naydenova Z, Hanna WJ, Coe IR. 2009. Disrupted plasma membrane localization and loss of function reveal regions of human equilibrative nucleoside transporter 1 involved in structural integrity and activity. Biochim Biophys Acta 1788:2326–2334.
- Procino G, Carmosino M, Marin O, Brunati AM, Contri A, Pinna LA, Mannucci R, Nielsen S, Kwon TH, Svelto M, Valenti G. 2003. Ser-256 phosphorylation dynamics of Aquaporin 2 during maturation from the ER to the vesicular compartment in renal cells. FASEB J 17:1886–1888.
- Reyes G, Chalsev M, Nivillac NMI, Coe IR. 2011. Analysis of recombinant tagged equilibrative nucleoside transporter 1 (ENT1) expressed in E. coli. Biochem Cell Biol 89:246–255.
- Robillard KR, Bone DB, Park JS, Hammond JR. 2008. Characterization of mENT1Delta11, a novel alternative splice variant of the mouse equilibrative nucleoside transporter 1. Mol Pharmacol 74:264–273.
- Salazar C, Höfer T. 2009. Multisite protein phosphorylation – from molecular mechanisms to kinetic models. FEBS J 276:3177–3198.
- Saxena SK, Kaur S. 2006. Regulation of epithelial ion channels by Rab GTPases. Biochem Biophy Res Commun 351:582–587.
- Sen RP, Delicado EG, Alvarez A, Brocklebank AM, Wiley JS, Miras-Portugal MT. 1998. Flow cytometric studies of nucleoside transport regulation in single chromaffin cells. FEBS Lett 422:368–372.
- SenGupta DJ, Lum PY, Lai Y, Shubochkina E, Bakken AH, Schneider G, Unadkat JD. 2002. A single glycine mutation in the equilibrative nucleoside transporter gene, hENT1, alters nucleoside transport activity and sensitivity to nitrobenzylthioinosine. Biochemistry 41:1512–1519.
- SenGupta DJ, Unadkat JD. 2004. Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem Pharmacol 67:453–458.
- Seo J, Lee KJ. 2004. Post-translational modifications and their biological functions: Proteomic analysis and systematic approaches. J Biochem Mol Biol 37:35–44.
- Stolk M, Cooper E, Vilk G, Litchfield DW, Hammond JR. 2005. Subtype-specific regulation of equilibrative nucleoside transporters by protein kinase CK2. Biochem J 386:281–289.
- Sundaram M, Yao SY, Ingram JC, Berry ZA, Abidi F, Cass CE, Baldwin SA, Young JD. 2001. Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J Biol Chem 276:45270–45275.
- Torres GE. 2006. The dopamine transporter proteome. J Neurochem 97:3–10.
- van Balkom BW, Savelkoul PJ, Markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PM. 2002. The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J Biol Chem 277:41473–41479.
- Wang JQ, Liu X, Zhang G, Parelkar NK, Arora A, Haines M, Fibuch EE, Mao L. 2006. Phosphorylation of glutamate receptors: A potential mechanism for the regulation of receptor function and psychostimulant action. J Neurosci Res 84:1621–1629.
- Weinman EJ, Steplock D, Zhang Y, Biswas R, Bloch RJ, Shenolikar S. 2010. Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J Biol Chem 285:25134–25138.
- Young JD, Yao SY, Sun L, Cass CE, Baldwin SA. 2008. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38:995–1021.