Abstract
Recent developments in the understanding of molecular diffusion phenomena in membranes are reviewed. Both model bilayers and biological membranes are considered in respect of lateral diffusion, rotational diffusion and transverse diffusion (flip-flop). For model systems, particular attention is paid to recent data obtained using surface-specific techniques such as sum frequency generation vibrational spectroscopy on supported lipid bilayers, and fluorescence correlation spectroscopy on giant unilamellar vesicles, both of which have yielded new insights into the intrinsic rates of diffusion and the energetic barriers to processes such as lipid flip-flop. Advances in single-molecule and many-molecule fluorescence methodologies have enabled the observation of processes such as anomalous diffusion for some membrane species in biological membranes. These are discussed in terms of new models for the role of membrane interactions with the cytoskeleton, the effects of molecular crowding in membranes, and the formation of lipid rafts. The diffusion of peptides, proteins and lipids is considered, particularly in relation to the means by which antimicrobial peptide activity may be rationalized in terms of membrane poration and lipid flip-flop.
Introduction
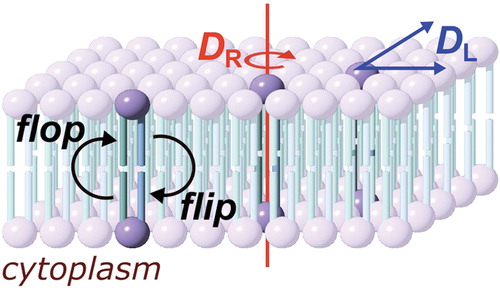
Lipid membranes are a fundamental component of biological systems, constituting both the barriers that maintain cell integrity and the partitions by which cells are divided into compartments with distinct biological and physiological properties. Our view of the biological membrane has changed significantly since the fluid mosaic model was developed by Singer and Nicolson (Singer and Nicolson Citation1972). Whilst the salient features of their model hold true, such as the innate ability of phospholipids to form bilayers and the potential for membrane proteins to diffuse within the bilayer, many of the now commonly recognized features of biological membranes are absent, including the inhomogeneous distribution of lipids and the presence of essentially static proteins that are associated with the cytoskeleton. The majority of biological membranes are a chemically diverse cocktail of lipids and proteins, with a broad range of lipid headgroups and acyl chains, alongside proteins that may constitute greater than 50% of the mass of the membrane. Membranes are frequently asymmetric, with non-uniform distributions of proteins and lipids both between leaflets and within a single leaflet. This asymmetry reflects the intrinsically dynamic nature of the membrane. Components are continually moving within the bilayer, forming complexes that may be long-lived, such as protein-protein adducts, or relatively short lived, such as protein-lipid or lipid-lipid adducts. Molecules are continually being recruited to or lost from the membrane, either as part of normal physiological processes such as signalling, or during recycling of membrane components. Understanding the rates by which peptides and proteins move within the membrane is therefore of fundamental importance for understanding a number of processes, including cell signalling, membrane poration, membrane fusion and the formation of lateral heterogeneity such as lipid rafts. Three general molecular processes can be described for membrane proteins and lipids: lateral diffusion in the plane of the membrane, rotation, and translocation between membrane leaflets. Other processes, such as molecular rocking or wobbling motions (Pastor et al. Citation2002, Pu et al. Citation2009) may also be considered, but have been less widely studied. Lateral diffusion, rotation and translocation are characterized respectively by the diffusion coefficient (DL), the rotational coefficient (DR) and the half-life for translocation (t1/2) (). For each of these processes, this review summarizes recent developments in the methodologies available for the study of their kinetics and the insights that are emerging from the use of these techniques. Model systems are widely used to understand the fundamental aspects of membrane kinetics. The key advantage of model systems is that most of the complexity inherent to biological membranes is simplified or removed, with the consequence that important principles governing membrane activity can be revealed systematically. This review will cover both the fundamental aspects of membrane kinetics revealed using model systems and how these aid our understanding of kinetic processes in biological systems.
Figure 1. Diffusion processes of membrane molecules: transverse diffusion (interleaflet exchange; flip-flop), lateral diffusion (characterized by the coefficient DL) and rotational diffusion (characterized by the coefficient DR). This Figure is reproduced in color in Molecular Membrane Biology online.
Lateral diffusion
Constraining factors for performing meaningful measurements on lateral diffusion rates are the ability to obtain data with sufficient temporal resolution to capture all diffusion phenomena, and the effects of labels introduced to facilitate spectroscopic measurements, which may perturb membrane fluidity, in terms of both bulk membrane viscosity (fluidity being inversely propotional to viscosity) and the lateral diffusion of individual components. A number of enhanced nanoscopy methods have been used to study the distribution of components in the lipid membrane and have been well reviewed (Duggan et al. Citation2008). These are not generally applicable to quantifying lipid dynamics, and are therefore not covered in detail here. Some techniques, most notably high-speed AFM, have not yet been widely adopted for quantifying diffusion within the membrane, but are likely to become increasingly important (Fantner et al. Citation2010, Casuso et al. Citation2011). Methods for studying lateral diffusion in the membrane have been reviewed (Kusumi et al. Citation2010, Owen et al. Citation2010) and the salient features of the principal methods used in the literature will be discussed first.
Model systems and methods for studying lateral diffusion
Supported lipid bilayers (SLBs)
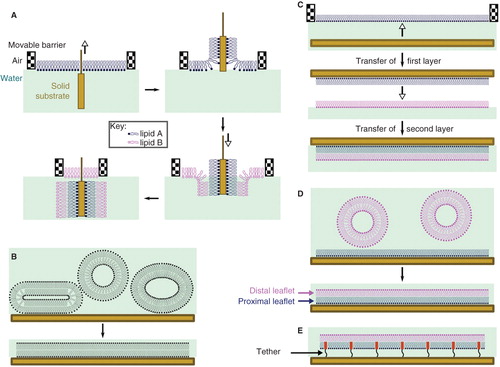
SLBs consist of a lipid bilayer adsorbed on the surface of a suitable solid substrate, such as gold, mica or silicon dioxide (Castellana and Cremer Citation2006, Czolkos et al. Citation2011), and are finding increasing usage both for studying lateral diffusion and interleaflet exchange of lipids. The choice of substrate and deposition method is determined by the requirements of the experiment in question. Three methods are generally used for preparing SLBs: Langmuir-Blodgett (LB, ) or Langmuir-Schaeffer (LS, ) deposition, vesicle fusion (VF, ), and a hybrid of the two (LS/VF, ), where vesicles are fused onto an existing monolayer. LB deposition involves the transfer of successive lipid monolayers from a suitable interface (e.g., air/water) to the solid surface by drawing the surface through the interface, and is well-suited to the preparation of model asymmetric membranes. The key advantages of this method are that the composition of each layer is easily controlled and deposition can be conducted at a controlled surface pressure, giving access to the fundamental thermodynamic parameters associated with lipid translocation. VF involves treating the surface with a liposome preparation and allowing the vesicles to fuse on the surface to form a complete layer. Whilst this method is the most convenient, it does not readily facilitate the formation of asymmetric bilayers unless combined with LS deposition of a single monolayer. It can be argued that the nature of the SLB, where one leaflet is closely associated with the solid support, is not a good representation of a biological membrane. However, neutron reflectometry experiments reveal that a 10–20 Å water layer containing ions remains trapped between the surface of the lipid layer and the solid support, and lateral mobility in the proximal leaflet is preserved (Johnson et al. Citation1991), indicating that the bilayer retains many of the properties of a free membrane. The spontaneous generation of lateral asymmetry in a bilayer formed by vesicle fusion has recently been reported (Wacklin Citation2011), presumably due to the specific interactions of some lipids with the surface, with the consequence that the chemical identity of each of the membrane leaflets cannot be assumed with SLBs prepared in this manner. In part because of concerns about the interaction of the proximal leaflet with the substrate surface, the use of a polymer cushion between the solid support and the bilayer has become more common (), providing for a greater water layer depth (Castellana and Cremer Citation2006, Czolkos et al. Citation2011).
Figure 2. Methods for the preparation of supported lipid bilayers (SLBs). (A) In the Langmuir-Blodgett (LB) method, the solid substrate is drawn through a monolayer of one lipid (lipid A) and subsequently pushed through a second layer (lipid B), producing an asymmetric layer. Each monolayer is at a controlled area per lipid molecule and surface pressure, giving excellent control of the composition of the SLB; (B) Vesicle fusion (VF) is the simplest method, but is not useful for the preparation of asymmetric bilayers; (C) In the Langmuir-Schaeffer (LS) method, entire intact monolayers are transferred to the solid substrate in successive operations; (D) Hybrid LS/VF or LB/VF methods. These allow asymmetric bilayers to be prepared in situ and are ideal for conducting measurements on SLBs immediately after preparation; (E) Tethered or polymer-supported bilayers consist of an amphiphile anchored to the surface of the substrate by a polymer (e.g., polyethyleneglycol), around which the proximal monolayer is formed. Tethered bilayers have a greater water layer depth between the proximal surface of the bilayer and the solid support. This Figure is reproduced in color in Molecular Membrane Biology online.
Fluorescence recovery after photobleaching (FRAP)
FRAP requires the membrane to be labeled with a suitable fluorophore. An incident beam of laser light is focused on the membrane, leading to photobleaching of the fluorophore within the focal spot. The rate of diffusion of non-bleached fluorophores into the bleached area is then monitored. The main drawbacks with this method are that photobleaching is not instantaneous and may not always be fully irreversible, with the consequence that some molecules will diffuse into the spot during the bleaching process (Weiss Citation2004). These factors, together with the flickering behavior typical of some fluorophores, introduce error into the measurements (Periasamy et al. Citation1996). Improved accuracy can be achieved by using spots of varying size to account for marker ingress during photobleaching, using pulsed lasers (van den Bogaart et al. Citation2007), or through the use of laser beams with modified profiles (Berkovich et al. Citation2011). Two-color methods, demonstrated using the photoconvertible protein dendra2, offer the possibility of simultaneously monitoring marker ingress during fluorescence recovery (of the green form of the dendra2) and marker egress from the irradiated area (of the red form of the dendra2), providing useful controls for modelling the data (Kaya et al. Citation2011).
Single fluorescent molecule tracking (SFMT)
Historically, monitoring the movement of a single molecule bearing a fluorescent marker was challenging, principally because the period in which the fluorophore could be tracked before photobleaching occurred was short and detectors lacked the sensitivity required. More recently however, the advent of new chromophores, allied to improvements in detector sensitivity, has made SFMT a feasible process (Kusumi et al. Citation2010, Citation2011, Rolfe et al. Citation2011). A key requirement for SFMT is to track the movement of single molecules with a high enough frame rate to capture sufficient data for analysis (Skaug et al. Citation2011). Frame rates of 10–1000 per second are typical, with a spatial resolution of 30–40 nm.
Single particle tracking (SPT)
SPT is typically conducted using either colloidal gold particles of diameter 20–100 nm (Eisenthal Citation2006), or quantum dots (QDs) of 2–10 nm diameter, composed of CdSe or ZnS (Biju et al. Citation2010, Pinaud et al. Citation2010). Methods involving gold particles rely on the scattering of incident light to generate interference contrast, producing high signal-to-noise ratios and excellent spatial resolution (2–20 nm, depending on frame rate). QDs are fluorescent, with the emission wavelength tuneable according to their diameter providing opportunities for multi-color labeling (Roullier et al. Citation2009). Importantly, QDs are very photostable. The movement of a single lipid or protein labeled with a QD can be tracked over long time periods (seconds to minutes) and large length scales (tens of micrometers). QDs are large in relation to the molecules to which they are attached. It has been estimated that attachment of a quantum dot is equivalent to adding an extra protein domain of ∼500 kDa to the molecule (Schneider et al. Citation1998). This will have some effect on the rate of lateral diffusion, although the 2-dimensional diffusion of membrane proteins is modelled well by Saffman-Delbrück theory (Saffman and Delbrück Citation1975, Mika and Poolman Citation2011), in which the diffusion coefficient varies according to the logarithm of the inverse of the radius of the section of the protein embedded within the membrane (i.e., DL ∝ ln(1/R), where R is the radius of the embedded section of the protein). As a consequence, protein dimerization for example, will produce a change in DL of ∼ 10%. Furthermore, as the viscosity of the membrane is significantly higher than the bulk medium (water), the attachment of a large water-exposed group to the membrane protein only has a small effect on DL. In some cases, attachment of QDs to transmembrane proteins has been found to produce anomalous diffusion (Nechyporuk-Zloy et al. Citation2008). Furthermore, in some cases QDs have demonstrated a tendency to self-associate to form clusters (Kusumi et al. Citation2010). There is therefore some potential for modification with a QD to hinder the diffusion of the labeled molecule. As with SFMT, the frame rate has an important influence on the quality of the data obtained, as low frame rates cannot capture localized variations in diffusion.
Fluorescence correlation spectroscopy (FCS)
FCS measures the fluorescence intensity in the focal spot of a laser as a function of time. From the autocorrelation function (Noda Citation2010), the fluorophore density and residence time in the spot are determined. FCS, like FRAP, is a many molecule technique, requiring data to be averaged for a large number of molecules to achieve good signal-to-noise ratios (Kusumi et al. Citation2010, Melo et al. Citation2011). In FCS, the minimum size of the focal spot (∼200 nm) limits the spatial resolution, which has led to the development of STED-FCS, in which the size of the focal spot is reduced below the optical diffraction limit to ∼30 nm. This in turn leads to improved modelling of the data to extract lateral diffusion coefficients (Mueller et al. Citation2011).
Pulsed field gradient nuclear magnetic resonance spectroscopy (PFG-NMR)
This method is commonly used for probing dynamics in model lipid systems such as liposomes. Unlike the methods described above, this technique can be performed without the introduction of labels, and in ideal cases can distinguish individual components of the membrane, making it a valuable analytical method. The membranes to be studied need to be aligned with respect to the experimental frame of reference, which may be achieved using bilayers formed on glass plates, or by using magnetically aligned bicelles (Horst et al. Citation2011, Macdonald and Soong Citation2011). During the signal acquisition sequence, two pulsed magnetic fields are applied at different times that lead to changes of intensity for molecules that have moved between the pulses, with the magnitude of the change dependent on the extent to which molecules have moved, i.e., DL. Limitations of this approach include the line broadening associated with the NMR spectra of lipid membranes, which may require the experiments to be conducted at the magic angle and limits the resolution that can be obtained, and the difficulty in distinguishing membrane components such as cholesterol that only have 13C and 1H nuclei available.
Measurements of DL in model systems
Single component lipid systems
For simple membranes formed from single lipid components in the fluid phase at room temperature, such as DOPC or POPC, values for DL in the range 5–8 μm2 s-1 are typical when determined by FCS methods on giant unilamellar vesicles (GUVs), regardless of the fluorescent species that is monitored (Kahya et al. Citation2003, Kahya and Schwille Citation2006, Przybylo et al. Citation2006, Ariola et al. Citation2009). Similar values are obtained by PFG-NMR (Orädd et al. Citation2002, Filippov et al. Citation2003, Lindblom et al. Citation2006). However, DL values obtained by FCS on supported lipid bilayers (SLBs) tend to be lower by a factor of 2–5 when compared with free-standing vesicle membranes of similar composition, even for SLBs separated from the surface by a polymer cushion (Sonnleitner et al. Citation1999, Zhang and Granick Citation2005). In most cases, the differences in DL between the bilayer leaflets on SLBs are small and within experimental error (5–10%), regardless of whether the SLB is formed directly on the solid support or on a polymer cushion (Wagner and Tamm Citation2000, Naumann et al. Citation2002, Zhang and Granick Citation2005), although in some cases DL values for the proximal and distal leaflet have been reported to differ by an order of magnitude (Hennig et al. Citation2009). The lower DL values for SLBs formed directly on a solid surface may be taken as an indication of a frictional interaction between the proximal (inner) leaflet and the surface, with similar DL values for both leaflets of SLBs suggestive of frictional coupling between the leaflets (Przybylo et al. Citation2006).
As expected, diffusion coefficients increase as the temperature is raised, with DL values of 20–30 μm2 s-1 reported for DMPC, DPPC, DOPC and POPC using PFG-NMR at 60°C (Filippov et al. Citation2003, Lindblom et al. Citation2006). Significantly slower diffusion coefficients are found for sphingomyelin (SM) membranes at room temperature, with values < 0.5 μm2 s-1, consistent with these membranes existing in the gel state at this temperature (Kahya et al. Citation2003, Ariola et al. Citation2009). Diffusion coefficients are higher in SM membranes at increased temperatures, although still considerably slower than fluid PC membranes, with values in the range 2–4 μm2 s-1 at 40–42°C and 9–12 μm2 s-1 at 60°C (Filippov et al. Citation2003, Lindblom et al. Citation2006). Studies of multilamellar liposomal membranes by neutron scattering (Busch et al. Citation2010) and simulation (Falck et al. Citation2008) have found evidence for flow-like behavior, in which lipid molecules move collectively. It remains to be seen if this is a general phenomenon that needs to be considered when accounting for differences in lateral diffusion rates between model and biological membranes, particularly as long-range flow is likely to be restricted by interactions of the cell membrane with the cytoskeleton.
Binary and ternary lipid mixtures
Complex mixing behavior is found for model membranes consisting of a ternary mixture of a lipid with a high gel to liquid crystal phase transition temperature (high-Tm lipid), a lipid with a low gel to liquid crystal phase transition temperature (low-Tm lipid) and cholesterol. Under appropriate conditions of temperature and composition, these membranes separate into macroscopic fluid liquid-disordered (ld) and condensed liquid-ordered (lo) domains (Veatch and Keller Citation2005, Honerkamp-Smith et al. Citation2009). The lo domain is enriched in cholesterol and the high-Tm lipid, with the low-Tm lipid localized predominantly in the ld domain. Although this macroscopic phase separation does not occur in vivo, the lipids involved in the formation of lo domains in vitro are frequently isolated from detergent-resistant extracts of biogenic membranes (Lagerholm et al. Citation2005, Lichtenberg et al. Citation2005, Brown Citation2006), and the same lipids are proposed to be components of lipid rafts. For the purposes of this review, lipid rafts are defined as localized regions of heterogeneity in biological membranes that form dynamically on a small scale (diameter ≤40 nm) (Lingwood and Simons Citation2010). It is therefore of intrinsic interest to quantify diffusion coefficients in model systems that exhibit domain formation. Ternary mixtures of DOPC, SM and cholesterol have been studied extensively by PFG-NMR and FCS. For mixtures with a DOPC/SM/cholesterol composition that is close to 1:1:1, at temperatures below 25°C, two separate diffusion constants are obtained for the lo and ld phases, with DL values of 0.2–0.8 μm2 s-1 and 3–6 μm2 s-1, respectively (Kahya et al. Citation2003, Lindblom et al. Citation2006, Ulrich et al. Citation2008). These values are in broad agreement with the diffusion coefficients for single component fluid and gel phase membranes, as well as binary mixtures of SM/cholesterol (1:1), which are in an lo phase at cholesterol concentrations > 35 mol% (Filippov et al. Citation2003, Kahya and Schwille Citation2006). As the temperature of the DOPC/SM/cholesterol ternary system is increased, the ld and lo phases coalesce to form a single phase with a diffusion coefficient (14 μm2 s-1 at 60°C) that is intermediate between those of fluid and gel phase membranes at the same temperature (Lindblom et al. Citation2006). Comparable diffusion coefficients for ld and lo phases have been obtained following microphase separation of ternary DOPC/DPPC/cholesterol mixtures (Lindblom et al. Citation2006); in this case the lo phase is enriched in DPPC and cholesterol. Binary mixtures of DPPC/cholesterol similarly exhibit diffusion properties typical of an lo phase (0.8 μm2 s-1 at 24°C) (Lindblom et al. Citation2006). Through the use of deuterium-labeled DPPC in binary mixtures with cholesterol it has been possible to determine separate DL values for each of the components over a range of temperatures and compositions (Scheidt et al. Citation2005). In these binary mixtures, cholesterol concentrations < 35 mol% yield complex mixtures of lo, ld and solid-ordered (so) phases, whereas cholesterol concentrations > 35 mol% produce pure lo phases. For most of these phases, including lo, the diffusion coefficients of each component are found to follow similar trends, with cholesterol always diffusing slightly faster than DPPC. Similar effects have been observed using 19F-labeled cholesterol and have been attributed in part to an interaction between the lipid and cholesterol, and in part to the lower molecular weight of cholesterol with respect to the lipid (Orädd et al. Citation2002). As a general rule, diffusion coefficients decrease with increasing cholesterol content in membranes containing cholesterol and a single low-Tm lipid. By contrast, diffusion coefficients are increased in binary mixtures with a high-Tm lipid as the cholesterol content is raised (Filippov et al. Citation2003, Kahya and Schwille Citation2006, Day and Kenworthy Citation2009).
Peptides and proteins
Studies that have employed labeled peptides or proteins on SLBs or GUV membranes have revealed interesting details of the effects of bilayer structure on lateral diffusion. A study on the effects of membrane curvature on the lateral diffusion of lipids and the potassium channel KvAP was conducted using SPT with quantum dots (Domanov et al. Citation2011). This yielded slower diffusion rates for the protein (DL = 2.3 μm2 s-1) when compared with the lipid (DL = 3.3 μm2 s-1), in accordance with Saffman-Delbrück theory (Saffman and Delbrück Citation1975). This study also demonstrated that DL is inherently sensitive to the curvature of the membrane, with diffusion coefficients increasing in proportion to the logarithm of the diameter of a membrane tube pulled from the surface of a GUV. Experiments to probe the effects of hydrophobic mismatch on the lateral diffusion of a transmembrane peptide have revealed that peptide mobility in GUVs formed from SOPC is greatest (DL ∼ 0.4 μm2 s-1) when the length of the hydrophobic transmembrane segment matches the hydrophobic thickness of the bilayer (Gambin et al. Citation2010). Lipid diffusion coefficients in these experiments were in line with those described above for fluid phase lipids (DL ∼ 5 μm2 s-1). Faster peptide diffusion was also obtained when the bilayer was of sufficient thickness that contact was reduced between a (non-transmembrane) peptide embedded in one monolayer and the lipids of the other monolayer (Gambin et al. Citation2010).
Incorporation of an amphipathic peptide fragment of hepatitis C virus non-structural protein 5A into an SLB formed from POPC was found to reduce the lipid diffusion coefficient from 2.0 μm2 s-1 to almost zero when studied by FRAP. Control experiments with a similar peptide of reduced amphipathicity yielded normal diffusion rates. On the basis of atomic force microscopy this reduced lateral mobility was attributed to membrane thinning induced by the peptide (Cho et al. Citation2007).
Measurements of DL in biogenic membranes
Diffusion coefficients measured in vivo, for both lipids and proteins, are typically an order of magnitude smaller than those determined using model systems. For lipids, DL is typically in the range 0.1–0.9 μm2 s-1 (Crane and Verkman Citation2008, Golebiewska et al. Citation2008, Citation2011, Baier et al. Citation2010, Mueller et al. Citation2011), with the corresponding values for peptides and proteins covering a greater range of 0.001–0.2 μm2 s-1 in most cases (Crane and Verkman Citation2008, Roullier et al. Citation2009, Baier et al. Citation2010, Won et al. Citation2010, Kaya et al. Citation2011, Valentine and Haggie Citation2011). These reduced diffusion rates have been attributed to two fundamental causes (Dix and Verkman Citation2008, Mika and Poolman Citation2011): (i) Molecular crowding; and (ii) membrane-cytoskeletal interactions. Molecular crowding is a reflection of the high protein content of most biogenic membranes, with integral membrane proteins occupying > 20% of the area and ∼ 20% of the mass of the plasma membrane (Dupuy and Engelman Citation2008), extending to ≥50% of the mass in the inner mitochondrial membrane (Zinser et al. Citation1991). Molecular motion in these crowded membranes may therefore be restricted by the high protein content. Consistent with this theory, it has been demonstrated that DL decreases linearly with respect to increasing protein concentration in GUV membranes (Ramadurai et al. Citation2009) and SLBs (Horton et al. Citation2010). Membrane-embedded proteins that interact with the cytoskeleton naturally display restricted lateral diffusion (Haggie et al. Citation2006, Crane et al. Citation2008, Valentine and Haggie Citation2011). Membrane proteins anchored to the cytoskeleton have been implicated in the generation of anomalous diffusion patterns for other membrane molecules.
Anomalous diffusion
Recent experiments in biogenic membranes have yielded complex patterns of lateral diffusion. In normal (Brownian) diffusion, the mean square displacement (MSD) of molecules increases linearly with regard to the length of the observation period: if the period of observation is doubled, the MSD also doubles. However, in some cases, the MSD of membrane components has been found to increase (superdiffusion) or decrease (subdiffusion) if the length of the observation window is increased (Dix and Verkman Citation2008), both of which may be described as examples of anomalous diffusion. In SPT experiments, anomalous diffusion is manifested by localized variations in the mean square displacement (Calvo-Muñoz et al. Citation2011). When monitored at low frequency, molecules appear to diffuse normally. In contrast, when monitored for sufficient periods at high frequency, the trajectories are divided in to small localized regions (diameter ∼30–300 nm) within which diffusion is normal (DL = 0.1–0.6 μm2 s-1 for lipids and transmembrane proteins at 37°C), with relatively infrequent ‘hops’ between adjacent regions (Kusumi et al. Citation2010, Citation2011, Crane et al. Citation2010, Valentine and Haggie Citation2011). The hop diffusion rate is the measured DL (typically ≤0.01 μm2 s-1) when the sampling frequency of the trajectory is low. Slow hop diffusion between regions (compartments), within which diffusion is relatively fast, has been accounted for by picket-fence models, which include transmembrane proteins anchored to the cytoskeleton as the ‘pickets’ and membrane cytoskeletal proteins such as actin filaments as the ‘fence’ (Golebiewska et al. Citation2011, Kusumi et al. Citation2011). Lipid rafts may also account for some instances of anomalous diffusion and it is notable that the compartment size in the picket-fence models are of the same order of magnitude as lipid rafts (Kusumi et al. Citation2010, Citation2011). Anomalous diffusion is not a ubiquitous phenomenon. For example, the diffusion properties of some proteins, such as aquaporin-1 (Crane and Verkman Citation2008) and the nicotinic acetylcholine receptor (Baier et al. Citation2010), are normal and unchanged following actin depolymerization by latrunculin (Frick et al. Citation2007). Aquaporin-4 on the other hand, shows more complex behavior in COS-7 cells, with the M1 isoform showing normal diffusion and the M23 isoform, which is able to assemble into slowly diffusing orthogonal arrays of particles, showing anomalous diffusion (Crane et al. Citation2010). Normal diffusion has also been reported for a number of G-protein coupled receptors (Kaya et al. Citation2011). As one would expect, the lateral diffusion behavior of a membrane protein is specific both to the cell in question and the region on the cell surface where the measurement is obtained. For example, QD-labeled BKCa channels show anomalous diffusion in COS-7 cells and the somatal and axiodendritic regions of neuronal cells, with the diffusion coefficients differing by an order of magnitude (Won et al. Citation2010). In the ER, inositol 1,4,5-trisphosphate receptors have been shown to display differences in mobility and distribution according to subtype (Pantazaka and Taylor Citation2011). Lipidated proteins that are associated with detergent-resistant membrane extracts have been shown to change from anomalous diffusion to normal diffusion upon cholesterol depletion of the membrane, suggesting a role for cholesterol in modifying diffusive behavior (Delint-Ramirez et al. Citation2011).
Lipid rafts
As described above, under appropriate conditions in model systems, macroscopic lo domains are formed that are enriched in cholesterol and a high-Tm lipid. In biogenic membranes however, macroscopic domains are not observed and the distributions of components that are commonly associated with rafts, such as GPI-anchored proteins and SM, appear to be homogeneous when studied by microscopy (Jacobson et al. Citation2007). Rather, domains enriched in SM and cholesterol form dynamically on a scale (<40 nm) that is smaller than the optical diffraction limit (Hancock Citation2006, Jacobson et al. Citation2007, Lingwood and Simons Citation2010). These smaller domains frequently contain transmembrane, GPI-anchored or lipidated proteins, and are ‘primed’ to coalesce into larger domains during cell signalling (Lingwood and Simons Citation2010) or under the influence of membrane tension (Ayuyan and Cohen Citation2008). It is notable that this scale is also smaller than the size of the compartments in the picket-fence model. Much of the evidence for raft formation arises from the partitioning and diffusion behavior of probes. The choice of probe is not always trivial; for example, many labeled SM and cholesterol analogs partition into ld rather than lo domains due to the steric bulk of the modification (Wang and Silvius Citation2000, Shaw et al. Citation2006, Baumgart et al. Citation2007, Loura et al. Citation2009). In some cases the properties revealed by a probe, such as slow diffusion, may arise from the effects of anomalous diffusion combined with a sample rate that is too slow (for SPT) or inappropriate modelling of data (for FCS) (Kusumi et al. Citation2010). The recent application of STED-FCS to the study of lipid dynamics within plasma membranes has led to improved spatial resolution and better modelling of the diffusion process (Mueller et al. Citation2011). This work accounted for the potential of probe modifications to modify partitioning behavior through the deployment of lipids labeled in either the headgroup of the acyl chain and highlighted the distinction between long-chain saturated lipids that diffuse slowly (in relative terms) and short-chain or unsaturated lipids that diffuse more rapidly. SM presented anomalous diffusion in the presence of cholesterol and normal diffusion following cholesterol depletion, with the rate of SM diffusion increasing in response to the reduction in cholesterol levels. Normal diffusion of SM was also observed after treatment with latrunculin B to induce actin depolymerization. Lipids bearing hydroxyl groups (gangliosides, PI) displayed an increased tendency to self-associate, but this was independent of cholesterol.
Overall, a picture is emerging in which small raft domains form dynamically in biogenic membranes through differences in the rate of association and dissociation of specific lipid types. Whether lipid diffusion between compartments is related to the formation of rafts is the subject of some debate (Kusumi et al. Citation2010, Citation2011, Mueller et al. Citation2011). A major role of cholesterol appears to be the regulation of membrane fluidity and lipid dynamics (Owen et al. Citation2010), respectively, retarding or accelerating the lateral diffusion of unsaturated and saturated lipids, as seen for PFG-NMR studies with binary cholesterol-PC mixtures (Filippov et al. Citation2003, Kahya et al. Citation2003, Kahya and Schwille Citation2006, Day and Kenworthy Citation2009).
Rotational diffusion
In contrast to lateral diffusion, the rotational diffusion coefficient varies according to the inverse of the square of the radius of the embedded section of the protein, (i.e., DR ∝ 1/R2) (Saffman and Delbrück Citation1975). As a consequence, DR is a sensitive parameter for characterizing protein aggregation and protein-lipid interactions (Fooksman et al. Citation2007).
Methods for determining DR
The most commonly used methods in the literature are ESR (Marsh Citation2008, Ryba and Marsh Citation1992, Mainali et al. Citation2011) and polarized optical methods (Swaminathan et al. Citation1997, Peters and Cherry Citation1982, Fooksman et al. Citation2007, Yengo and Berger Citation2010). NMR is useful for molecules that rotate relatively slowly, such as peptides and proteins (Salnikov et al. Citation2010). A fundamental concern with determination of DR is whether any probes that have been introduced to a lipid or protein are reporting the behavior of the molecule as a whole, or localized torsional rotations. For this reason, in many cases DR measurements are made with the label incorporated at different positions in the molecule, such as the headgroup and acyl chains for lipids. Rotations about both the axis parallel to the membrane normal and the axis perpendicular to the membrane normal may be considered (Ge and Freed Citation2011).
Typical values for membrane proteins and lipids
n model gel phase membranes, lipids have a relatively low rotational diffusion coefficient of the order of 106–107 s-1. The value of DR increases in more fluid membranes, with reported values ranging form 107 s-1 for ld phases (Ariola et al. Citation2009), to 109 s-1 in PC/PG/cholesterol membranes (Ge and Freed Citation2011). Membrane proteins generally yield rotational diffusion coefficients that are significantly reduced in comparison to lipids, with 104 s-1 being typical (Cherry and Godfrey Citation1981, Peters and Cherry Citation1982), although fast rotation (DR = 106 s-1) has been reported for some peptides (De Angelis et al. Citation2011). The rotational diffusion of peptides is reduced to almost zero in gel phase membranes (Cornell et al. Citation1988). From a fundamental perspective, measurements of DR for membrane proteins have yielded data both in support (Peters and Cherry Citation1982) and against (Ariola et al. Citation2009) Saffman-Delbrück theory descriptions of diffusion in membranes.
Interleaflet lipid translocation (flip-flop)
It is now well-established that the plasma membranes of most cells are asymmetric with regard to the lipid composition of the cytoplasmic (inner) and extracellular (outer) leaflets. For example, in many eukaryotic cells, SM is enriched in the outer leaflet, whereas PE, PI and PS are predominantly located in the inner leaflet. The transmembrane distribution of lipids has been well reviewed (Pomorski et al. Citation2001, Boon and Smith Citation2002, Sanyal and Menon Citation2009, Devaux and Herrmann Citation2012). Transmembrane lipid distributions in eukaryotes are regulated by three main classes of enzyme: Floppases facilitate the movement of lipids from the inner leaflet to the outer leaflet, flippases facilitate the reverse translocation from the outer to the inner leaflet, and scramblases promote translocation in both directions (Daleke Citation2003, Devaux and Herrmann Citation2012). The proteins involved in lipid transport have proved particularly challenging to characterize and there is still some debate as to whether flip-flop is an active (ATP-dependent) or passive (ATP-independent) process, or a combination of both. Some of the translocases involved in maintenance of asymmetry are ATP-dependent, most notably aminophospholipid translocases in the ER and P4-ATPases in the plasma membrane (both flippases) (Verhulst et al. Citation2012). However, ATP-independent lipid translocation activity has been reported for microsomal membrane preparations that have been incorporated into liposomes, indicating that in the ER, energy-independent pathways for lipid flip-flop operate (Menon and Herrmann Citation2012). Much of the historical literature on transmembrane asymmetry is predicated on the propensity for lipids in the outer (but not inner) leaflets of biological membranes to undergo chemical reactions, or exchange with lipid vesicles (Etemadi Citation1980). Lipid exchange has traditionally been performed using labeled membrane lipids and unlabeled liposomes (LUVs) under catalysis by phospholipid exchange proteins (Rothman and Dawidowicz Citation1975, Lenard and Rothman Citation1976, Rothman et al. Citation1976), with the amount of lipid transferred to the liposomes quantified using standard analytical methods. These approaches work for a range of lipid types, although careful controls are needed to account for the lipid selectivity of the exchange protein. Methods employing enzymes that have lipids as substrates, such as phospholipase A2, phospholipase C and sphingomyelinase offer improved selectivity (Boon and Smith Citation2002), but again careful controls are needed due to the potential for perturbation of the membrane as the reactions near completion (Wacklin et al. Citation2007). All of these methods work on the assumption that only the lipids in the outer leaflet are accessible, i.e., that the rate of flip-flop is slow with regard to the experimental timescale, and that the conditions of the experiment do not perturb asymmetry. This renders the study of flip-flop rates in membranes of fundamental significance. There are additional biological reasons for understanding the rate of flip-flop. In some organelles, such as the endoplasmic reticulum (ER), fast flip-flop is a key requirement for ensuring the availability of lipids that serve as substrates for protein modification (Devaux and Zachowski Citation1994, Sanyal and Menon Citation2009). ATP-independent translocation of most lipid types in these organelles occurs readily. By contrast, translocation of lipids in the plasma membrane exhibits greater lipid selectivity, which is allied to specific roles for some lipids in cell physiology, such as PI in secondary messenger signalling (Chakraborty et al. Citation2011), and PS appearance in the outer leaflet is a marker for apoptosis (Uchida et al. Citation1998).
The rate of flip-flop is conveniently expressed as the half-life for interleaflet transfer, t1/2. Values for t1/2 range from short (s to min) for biological membranes such as the ER, to long (min to h) for the membranes of liposomes. For many years, following the classic experiment by (Kornberg and McConnell Citation1971) using spin-labeled diacylphosphocholine analogs, which established t1/2 values in the range 1.5–6.5 h at 30°C in synthetic membranes, the rate of flip-flop was generally accepted to be slow in biological membranes. More recently however, a number of studies on both model and biological membranes, using an array of alternative labeling strategies and spectroscopic approaches, have yielded a body of data that challenge the concept that flip-flop is always an inherently slow process. Three fundamental issues arise when considering these studies: firstly, it is clear that careful consideration of the effects of introducing a label on the translocation rate is needed; secondly, care is needed in assessing whether the method used is capable of delivering sufficient temporal resolution to resolve fast kinetic events; thirdly, it should be recognized that translocation rates are sensitive to the method used to prepare the membrane under investigation. As a consequence of these factors, comparisons of different experimentally determined flip-flop rates are challenging to make, even where the same lipid system has been studied. The methods most commonly used in the recent literature are outlined below.
Model systems and methods for the determination of flip-flop rates
Sum frequency generation vibrational spectroscopy (SFVS)
Studies on SLBs have provided a rich body of data on the fundamental aspects of flip-flop, particularly when allied to SFVS. SFVS is a form of non-linear optical spectroscopy that provides similar information to Raman and infra-red spectroscopy (Liu and Conboy Citation2004, Citation2005, Eisenthal Citation2006, Yang et al. Citation2011). The method involves two lasers focused at the interface, one of which is operated at a fixed-wavelength in the UV range (ωuv), the other being scanned over the IR frequency range (ωir). These two combine to form a signal at the sum frequency (ωsum = ωuv + ωir). When the sources are polarized and the sum signal focused at an appropriate surface, the incident beam is able probe the orientation of surface molecules with sub-monolayer resolution. The output signals are in the IR frequency range and contain orientation information. When applied to bilayers adsorbed on the surface of a quartz crystal prism, signals from bilayer elements that are symmetric about the plane of the membrane cancel each other out, rendering SFVS a particularly sensitive technique for studying membrane asymmetry. A further advantage of SFVS is that, in favourable cases, no labeling is required. Where labeling is needed, unobtrusive deuterium labels suffice to distinguish the membrane leaflets.
Fluorescence-based methods
These have the key advantage of being sufficiently sensitive and rapid to give excellent temporal resolution. However, the requirement for a component of the membrane to be modified with a non-biogenic fluorophore is a disadvantage. Translocation rates determined using fluorogenic lipids only reliably report the translocation rate of the labeled lipid in question, and not other classes of lipids of the membrane in which they are located. Methods used specifically to address bilayer asymmetry include fluorescence inhibition, by quenching or oxidation of the fluorophore in one leaflet by the addition of exogenous agents to one side of the membrane (Eckford and Sharom Citation2010, Pomorski et al. Citation2001), time-resolved emission spectroscopy (TRES) (Horng et al. Citation1995, Volinsky et al. Citation2011), fluorescence lifetime (Kułakowska et al. Citation2010) and fluorescence-interference contrast microscopy (FICS) (Crane et al. Citation2005).
Specific binding
In some cases, proteins that interact specifically with a particular class of membrane lipid can be used probe the appearance of that lipid in the outside leaflet of a cell or vesicle. This is most readily demonstrated by annexin V and lactadherin, which bind selectively to PS (Metkar et al. Citation2011) and pleckstrin homology domains, which have binding selectivity for PI lipids (Hurley and Meyer Citation2001, Hurley Citation2006).
The debate over flip-flop: fast or slow?
The breakthrough work by Kornberg and McConnell established lipid exchange in vesicles with a t1/2 in the range of 0.7–3 h (at 40°C), with an activation energy barrier to translocation (Ea) in the range 65–116 kJ mol-1 (Kornberg and McConnell Citation1971). Since then, a plethora of papers have published t1/2 data for most lipid types, with values spanning a huge range, from ms to days. It is not the intention to reproduce all of these data here, but salient examples will be selected to highlight the issues with interpretation of the data, and give a sense of typical values for common lipid types.
A series of SFVS experiments by Conboy have determined the rates of flip-flop in PC membranes on silica surfaces. Determination of the effects of temperature on flip-flop rates has revealed some of the fundamental thermodynamic aspects of the process in these systems. In general, longer chain PCs are found to undergo flip-flop more slowly than shorter chain counterparts, with a value for Ea of ∼ 220 kJ mol-1 for DPPC being typical. The free energy barrier to translocation (ΔG‡) is typically of the order of 100 kJ mol-1 and includes a particularly large positive entropic component (TΔS‡ ≈130 kJ mol-1) (Liu and Conboy Citation2005, Anglin et al. Citation2010). Most strikingly, the rates of flip-flop in these experiments are significantly faster than observed in biological membranes, with t1/2 in the range of seconds to minutes, e.g., t1/2 = 52 min for DPPC at 25°C and 30 mN/m surface pressure. The rate of flip-flop was found to be the same in both directions and the bilayers exhibited normal lateral diffusion rates, appeasing to some extent concerns that interaction of the bilayer with the solid support was responsible for the fast translocation rates. However, similar experiments by SFVS using asymmetric DPPC/dDPPC bilayers at 24°C yielded little exchange over course of an hour (Yang et al. Citation2011). Studies using FICS on polymer-tethered SLBs composed of POPC at 22°C (Kiessling et al. Citation2006) also gave significantly larger values for t1/2 (15 h), which is particularly notable as flip-flop is expected be faster in the fluid phase POPC bilayer than the gel phase DPPC bilayer at this temperature. However, the lateral diffusion coefficient (DL) of labeled lipids was lower in this system than comparable examples with the same lipid composition. Of further note, a lipid with the same TEMPO spin label as the Kornberg and McConnell experiments was shown to undergo slower flip-flop in the Conboy SLB experiments than the same lipid when unmodified (Liu and Conboy Citation2005). This raises the question of whether the rate observed by Kornberg and McConnell is a true reflection of the rate of flip-flop in membranes, or represents an artifactually slower rate due to the presence of the lipid modification.
Another salient example of diverse t1/2 values has been provided by the examination of flip-flop rates in SLBs formed from DOPC/DOPS on ITO-treated glass (Kułakowska et al. Citation2010). Flip-flop in this system was addressed using fluorescent lipid analogs (Atto633-DOPE and F2N12S), with the variation in fluorescence lifetime (varying inversely with the distance from the ITO surface) used to assess flip-flop. Both of the labeled lipids gave similar and normal lateral diffusion rates (5–7 μm2 s-1), but flip-flop rates that differed by an order of magnitude according to the fluorescent lipid used (t1/2 values of 3 min for F2N12S and 32 min for Atto633-DOPE). Furthermore, F2N12S flip-flop in this system was significantly faster than found with the same label in biological membranes (t1/2 > 1 h).
Studies directed to assaying the flip-flop of PE in the presence of other lipids have demonstrated that flip-flop occurs readily for this lipid, at similar rates to PC (Anglin and Conboy Citation2009). Flip-flop of PS is also well established (Langer and Langosch Citation2011, Volinsky et al. Citation2011), including a salient example of PS externalization within a period of < 5 min on cytotoxic lymphocytes and mouse CD8 cells, following administration of the protein perforin at concentrations at which the protein associated with membranes as a monomer (Metkar et al. Citation2011). Equinatoxin II, a peptide toxin, was found to induce a similar PS externalization in the same cells. Of all the molecules found in eukaryotic membranes, cholesterol has produced the greatest variation in determined flip-flop rates. At the fast end of the spectrum, Müller and Herrmann used nitroxide-labeled sterols in order to probe flip-flop in synthetic vesicles and erythrocytes (Müller and Herrmann Citation2002). Reduction of the spin-label by ascorbic acid was used to assess label exposure on the external leaflet, in a manner reminiscent of the Kornberg and McConnell experiments. Significantly, for one of their analogs (a cholestane), exchange occurred at a faster rate than reduction, which only enabled an upper limit to be placed on the process (t1/2 < 0.5 s). Using NMR methods that combined the use of [3-13C]-cholesterol with paramagnetic metal ions added on one side of the membrane, a t1/2 of <2 ms was determined for cholesterol in 30 nm SUVs composed of POPC/POPA (Bruckner et al. Citation2009). Although this rapid exchange may be facilitated by curvature strain in these SUVs, these experiments nevertheless demonstrate the applicability of NMR methods to resolve fast exchange processes with minimally disruptive labels. At the other end of the timescale, a t1/2 of 200 min has been reported for cholesterol flip-flop in POPC/cholesterol and dPOPC/cholesterol liposome preparations at 50°C, using small-angle neutron scattering approaches (Garg et al. Citation2011). Using similar SANS methodology on PC liposomes at 37°C containing varying quantities of cholesterol, cholesterol has been shown to slow down PC flip-flop by several orders of magnitude, from 350 min in the absence of cholesterol to > 4.5 days in membranes with 40% cholesterol (Nakano et al. Citation2009). Under the conditions of these experiments, the PC components (DMPC or POPC) are expected to be in the fluid state at 37°C in the absence of cholesterol, and a liquid-ordered state at 40% cholesterol. The slower flip-flop rates are consistent with the formation of a more closely-packed membrane with a higher Ea towards exchange. Slow flip-flop rates are more in keeping with the historical exchange rates determined for biogenic membranes such as those of enveloped viruses, using PLD or phospholipid exchange experiments, where t1/2 values of > 13 days for cholesterol, >10 days for PC and > 30 days for SM have been reported (Lenard and Rothman Citation1976, Rothman et al. Citation1976).
Recent reports of the induction of asymmetry in bilayers on solid supports (Wacklin Citation2011) and in liposomal membranes following the administration of poly-L-lysine (Brown and Conboy Citation2011) or the peptides melittin and alamethicin (Qian and Heller Citation2011), shed a note of caution on the interpretation of flip-flop rates in biogenic membranes. In thermodynamic terms, these experiments demonstrate previous theories (reviewed in Boon and Smith Citation2002) that the free energy benefit from the electrostatic interaction of membrane lipids with a surface or a macromolecule can be sufficient to compensate for the increase in free energy associated with the establishment of membrane asymmetry. In terms of kinetics, the asymmetry in these systems is a dynamic equilibrium that reflects different t1/2 values for the separate exchange processes that occur in each direction in the bilayer. To put this in a more biological context, any lipid in the cytoplasmic leaflet of a membrane that has electrostatic interactions with a membrane protein will potentially exhibit a reduced rate of exchange to the extracellular leaflet (flop) compared to that of the reverse process (flip) and as a consequence demonstrate a preferential localization in the cytoplasmic leaflet. The actual extent of asymmetry in this scenario will be determined by the free energy released by forming electrostatic interactions between the protein and the membrane lipids. The rate of flip-flop however, only depends on the magnitude of the free energy gain needed to reach the transition state, ΔG‡. Therefore, in membranes with high asymmetry, the constituent components may still undergo fast interleaflet exchange. The ‘accessible’ component of the membrane in many studies of asymmetry, particularly for methods with low temporal resolution, may actually be reporting lipids from the extracellular leaflet and unbound lipids from the cytoplasmic leaflet. A particularly salient example is provided by the studies of viral envelope asymmetry described above that yield very large t1/2 values. As these viral envelopes are closely associated with a dense layer of matrix protein in close contact with inner surface of the envelope, it is reasonable to suspect that the reported asymmetry actually reflects interactions of specific lipids with the protein layer. From these arguments, it should be apparent that studies on flip-flop in biogenic membranes should aim to address lipid exchange in both directions with the highest possible temporal resolution.
Taken together, the available data indicate some general patterns: flip-flop in free-standing fluid membranes generally occurs with a t1/2 of the order of hours to days. Flip-flop in more condensed gel phase or liquid ordered membranes tends to be slower than in fluid phases. Flip-flop in SLBs formed directly on a solid-support is faster than in the equivalent membranes when not on a solid-support, or formed on a solid support with a polymer cushion. As will be discussed below, in some cases, particularly where the membrane is stressed, lipid exchange occurs much more rapidly.
Promotion of interleaflet exchange by peptides and proteins
Many of the studies of interleaflet exchange have been conducted using antimicrobial peptides and model peptides in vitro, but examples have also emerged of similar behavior with both peptides and proteins in vivo. The salient features of peptide-induced flip-flop are that it is both rapid and transient, occuring on a timescale of minutes following administration to the membrane (Fattal et al. Citation1994, Matsuzaki et al. Citation1996, Frasch et al. Citation2004). Slower translocation rates have been reported for integral transmembrane peptides (Kol et al. Citation2001, Citation2003). It should be noted that peptide-induced translocation is not a general phenomenon, particularly for transmembrane peptides (Marsh Citation2008). Three general mechanisms have been proposed for the increased flip-flop rates produced by peptides: (i) The formation of peptide-stabilized toroidal pores; (ii) membrane thinning induced by peptide binding; and (iii) flip-flop through membrane defects (Cho et al. Citation2007, Gurtovenko and Vattulainen Citation2007a, Anglin et al. Citation2009, Bocchinfuso et al. Citation2011, Fuertes et al. Citation2011, Salnikov and Bechinger Citation2011).
Toroidal pores
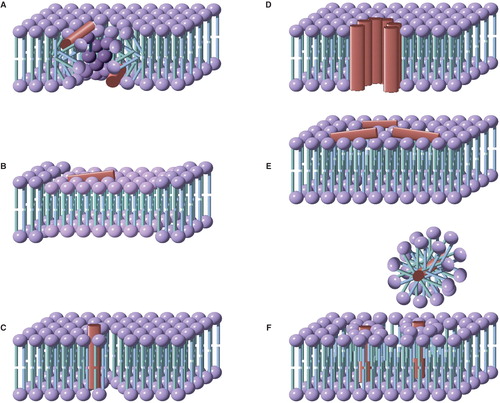
Peptide-induced flip-flop is frequently accompanied by poration of the membrane (i.e., release of ions or other markers), which has led to the development of models for poration that involve toroidal pores, in which the leaflets of the membrane become contiguous (), enabling rapid diffusive passage of lipids between leaflets. Pore formation occurs above a critical ratio of peptide to lipid (P/L*), and as a consequence the rate of flip-flop (and pore formation) is not linear with respect to peptide concentration (Huang Citation2006, Fuertes et al. Citation2011). The nature of toroidal pores implies that the energy barrier to flip-flop should be similar to that for diffusion in the plane of the membrane (Anglin et al. Citation2009), although a significant peptide density at the point of highest curvature facing the axis of the pore would be expected to hinder the rate of diffusion. Chen and co-workers used SFVS to demonstrate that that administration of the peptide MSI-78 to asymmetric dDPPG/DPPG supported bilayers led to a loss of asymmetry over a time period of >10 min following administration. Their data were interpreted in terms of a toroidal pore, as peptide concentrations greater then 2 μM were required to induce significant flip-flop (Yang et al. Citation2011).
Figure 3. Models for the mode of action of antimicrobial peptides. (A) Toroidal pores, stabilized by the peptide, consisting of a lipid-lined pore and contiguous membrane leaflets; (B) Membrane thinning, in which peptide binding leads to a reduction in the thickness of the bilayer. The thinner bilayer presents a reduced barrier to the flip-flop of lipids between leaflets, as well as to the formation of transient defects; (C) Defect-mediated poration, in which the formation of transient defects (that are significantly less hydrated than toroidal pores) is promoted following peptide binding; (D) The barrel-stave model, consisting of a peptide-lined pore; (E) The carpet model, in which bilayer integrity is disrupted by the formation of peptide aggregates; (F) Detergent models, in which membrane disruption occurs through the formation of peptide-lipid micelles that remove lipids from the membrane. This Figure is reproduced in color in Molecular Membrane Biology online.
Membrane thinning
Membrane thinning (i.e., a decrease in bilayer thickness, ) has been demonstrated in a number of cases following the association of peptides with membranes (Lee et al. Citation2005, Citation2008, Cho et al. Citation2007, Marsh Citation2008). Thinning may arise as a direct consequence of peptide association, or as a secondary effect resulting from changes in membrane curvature. In relation to the latter, molecular modelling data suggest that membrane thinning is a consequence of increased curvature (Risselada and Marrink Citation2009).
Conboy and co-workers have studied the induction of flip-flop by the model transmembrane helix WALP23 and the bee venom peptide melittin by SFVS on DSPC supported lipid bilayers (Anglin et al. Citation2009). Representative t1/2 values of 195 min at 36°C and 8.6 min at 34°C were obtained for DSPC + 1 mol% WALP23 and 1 mol% melittin, respectively, against a t1/2 of > 1 day in the absence of any peptide. These t1/2 values were in accord with the corresponding free energy barriers (ΔG‡) for flip-flop at 37°C of 107 kJ mol-1, 103 kJ mol-1 and 92 kJ mol-1 for DSPC, DSPC + 1 mol% WALP23 and DSPC + 1 mol% melittin, respectively. In the presence of WALP23, the relatively small change in ΔG‡ was comprised of greater but opposing changes in ΔH‡ and ΔS‡, with ΔH‡ being more favourable (smaller) by 30 kJ mol-1, and ΔS‡ less favourable, reflecting a more ordered transition state for flip-flop. In the case of melittin, the greater decrease in ΔG‡ for flip-flop produced by the peptide was almost entirely the result of changes in the entropic component of ΔG‡, with the enthalpic component not changing significantly. This corresponds to a more disordered transition state for flip-flop in the presence of melittin. For both peptides, the variation in ΔG‡ was linear in respect to peptide concentration, which does not fit with a toroidal pore model for flip-flop in this system. In addition, for both peptides, the enthalpic energy barrier to flip-flop was higher than would be expected for a toroidal pore. The data for melittin were therefore interpreted in terms of thinning of the membrane. These data are in accord with neutron scattering experiments that have shown that both melittin and the antimicrobial peptide alamethicin binding to the outer surface of a model membranes leads to membrane thinning via a reduction in thickness of the outer leaflet (Huang et al. Citation2004, Qian and Heller Citation2011). It should be noted that whilst melittin may also be implicated in the formation of toroidal pores (Yang et al. Citation2001, Huang Citation2006, Rapson et al. Citation2011), there have been other reports of increased flip-flop stimulated by melittin:lipid ratios below the critical peptide to lipid ratio for pore formation, in addition to the data from Conboy (Qian and Heller Citation2011). Membrane thinning has been reported for non-antimicrobial peptides, including a peptide from hepatitis C virus (Cho et al. Citation2007).
Defect-mediated poration
A key feature of this mechanism is the creation of membrane defects following peptide binding that permit flip-flop by lower energy pathways (). This mechanism has been proposed for a number of peptides, including alamethicin (Pabst et al. Citation2007), gramicidin A (Anglin et al. Citation2007), and the WALP23 peptide described above (Anglin et al. Citation2009). As the membrane is a dynamic structure, localized defects generated by thermal fluctuations will occur spontaneously and translocation through these is likely to be responsible for the background rate of translocation in the absence of peptides or other perturbing agents (Fuertes et al. Citation2011). Translocation through transient pores has been observed in simulations of bilayer systems (Tieleman and Marrink Citation2006, Gurtovenko and Vattulainen Citation2007a, Citation2007b, Marrink et al. Citation2009, Gurtovenko et al. Citation2010, Sapay et al. Citation2010). The formation of transient defects following peptide binding will potentially enable sufficient penetration of the hydrophobic region of the bilayer by water to reduce the degree of headgroup desolvation required during translocation. Both the membrane-thinning and the defect-mediated poration models yield translocation rates that increase approximately linearly in respect of peptide concentration. In many respects there is a large degree of synergy between the defect-mediated and membrane-thinning models. Association of molecules with the membrane interface in one leaflet of the bilayer is sufficient to increase the area of this leaflet, the ramifications of which are membrane thinning and a difference in surface tension between the leaflets that reduces the barrier to thermally-driven pore formation (Ludtke et al. Citation1995, Longo et al. Citation1998, Lee et al. Citation2008, Wimley Citation2010, Qian and Heller Citation2011). In common with the membrane thinning model, in defect-mediated poration the barrier to flip-flop is reduced, but the latter model is distinguished by more localized lipid disorder. The defect-mediated model may be further differentiated from both the toroidal pore and thinning models in having the faster apparent kinetics, primarily because membrane disorder is the immediate consequence of peptide binding. Added to this, solvent penetration of the bilayer, either following peptide-membrane binding or peptide desorption from the membrane (as peptide-membrane binding is intrinsically reversible), is expected to occur faster than lipid restructuring to anneal defects and lateral peptide diffusion during to drive assembly into toroidal pores. As a consequence, it is to be expected that in some experiments, complex translocation kinetics will be observed.
Other forms of membrane perturbation enhance the rate of translocation, including the process of heating or cooling a membrane through the gel to liquid crystalline phase transition (De Kruijff and Van Zoelen Citation1978). It is also of note that in both the paper by Kornberg and McConnell, and more recent studies (Kotova et al. Citation2011, Volinsky et al. Citation2011), the presence of oxidized lipids has been found to accelerate transbilayer exchange of membrane lipids. In both of the latter cases, packing defects generated by the oxidized lipids were proposed as the principal reason for translocation, although there are differences in the extent of water penetration of these defects in the two cases, ranging from little penetration (Kotova et al. Citation2011) to water-filled pores spanning both leaflets (Volinsky et al. Citation2011). Interestingly, when photodynamic methods are used to generate oxidized lipids (Kotova et al. Citation2011), the defects produced are sufficient to enable the passage of small organic molecules, but not ions, which is consistent with a sparsely hydrated pore. Most membranes in vivo are characterized by the existence of an electrochemical potential gradient, which may serve to drive the formation of more long-lived pores from transient defects (Gurtovenko and Vattulainen Citation2005, Citation2007a, Citation2007b, Gurtovenko et al. Citation2010). Whether a pore extending across the entirety of the bilayer is a required for lipid translocation is not certain; partial penetration of the hydrophobic interior of the bilayer by water will lead to similar effects to membrane thinning in terms of translocation rates.
Peptide-membrane binding kinetics
Models for peptide-membrane binding
A vast body of work has accrued on the subject of peptide interactions with membranes, much of it associated with understanding the mechanisms by which antimicrobial peptides operate. Models for antimicrobial peptide activity have been well reviewed (Huang et al. Citation2004, Reddy et al. Citation2004, Bechinger and Lohner Citation2006, Huang Citation2006, Shai 1999, Epand and Epand Citation2009, Wimley Citation2010). Salient models are summarized in . Historically, general models such as the barrel stave, toroidal pore and carpet models, as well as those involving detergent activity, have been peptide-centric, focused to a large degree on the peptide structures that form at equilibrium. In considering the process of peptide-membrane binding, understanding the kinetics of each stage of the process is essential, both with regard to the time that is required for equilibrium to be attained, and the timing of events such as marker release in relation to the equilibration process (Schwarz et al. Citation1987). Marker release following the administration of an active peptide is usually rapid and frequently involves biexponential kinetics, with the fast initial release on a timescale of seconds followed by a slower phase on a timescale of minutes to hours (Schwarz and Robert Citation1990, Citation1992, Mazzuca et al. Citation2010). In order to relate marker release kinetics to the binding process, it is necessary to consider a holistic approach that involves explicitly monitoring of the kinetics of both peptide and lipid processes within the mixture. Studies of this nature are infrequent; the majority of cases focus measurements on either peptide or lipid properties, with phenomena such as marker release usually implicitly interpreted in terms of peptide models. Recently, a more lipid-centric viewpoint has been called for that is more in keeping with the kinetics of poration phenomena (Fuertes et al. Citation2011). Before considering more lipid inclusive models, it is useful to consider alternative models for interpreting data from marker release experiments.
All-or-none or graded release
A fundamental issue with regard to antimicrobial peptide models concerns identification of the equilibrium structures that form. One of the methods that may be used to distinguish mechanisms that involve a discrete pore (barrel stave and toroidal pore) from those that involve transient poration (detergent-based and carpet model) is the nature of marker release observed from lipid vesicles containing entrapped markers (Schwarz and Robert Citation1990, Citation1992, Ladokhin et al. Citation1997, Arbuzova and Schwarz Citation1999). For all-or-none release, the formation of a stable long-lived pore at concentrations greater than a threshold concentration (P/L*) leads to rapid and complete release of entrapped markers from individual vesicles. The sample then comprises a mixture of completely empty vesicles and completely filled vesicles. Furthermore, the addition of a new marker to the solution outside the vesicles leads to marker ingress into porated vesicles only (Gregory et al. Citation2008, Citation2009).
By contrast, graded release is characterized by a slow release of markers over a finite time window following addition of the peptide, with the timescale for cessation of net marker release typically of the order of several minutes at non-saturating peptide concentrations (Pokorny and Almeida Citation2004, Gregory et al. Citation2009, Apellániz et al. Citation2010). The sample at this point comprises vesicles with varying states of partial emptiness. Addition of a new marker outside the vesicles does not lead to partial marker ingress into all vesicles. The recent wide adoption of giant unilamellar vesicles (GUVs) as model vesicular systems, combined with confocal microscopy, has provided some salient examples of all-or-none and graded poration. These experiments reveal details of the rate of marker influx or efflux once a long-lived pore has formed, as well as the time required for pore formation to occur. For example, two peptides from the HIV gp41 fusion protein were shown to differ in their mode of action in an assay that measured the rate of fluorescent marker influx into GUVs simultaneously treated with a peptide and the marker (Apellániz et al. Citation2010). One of the peptides, termed CpreTM, induced all-or-none poration of GUVs, characterized by fast filling kinetics, with GUVs reaching 80% filled within ∼ 100s. However, this rate of marker ingress was only achieved once a stable pore had formed. The rate of pore formation, as determined by the lag between adding the peptide and marker ingress starting, varied between 0 and 20 min across the sample. By contrast, the other peptide, termed NpreTM, exhibited a graded mechanism, with the extent of filling not reaching 30% after 30 h of incubation. This peptide also displayed a lag phase between peptide addition and poration commencing. Similar lag phases have been reported for marker release experiments using GUVs (Vad et al. Citation2010). Treatment of GUVs with the peptide Baxα5 led to the formation of stable pores, with some vesicles remaining permeable to markers after several hours, consistent with an all-or-none mechanism (Fuertes et al. Citation2010). Pores formed over longer time scales were smaller than the pores formed after initial peptide binding, suggesting that a rapid formation of non-equilibrium pores occurred alongside assembly to form stable pores. The differences between all-or-none and graded release revealed by GUV experiments may generally be interpreted in terms of equilibrium and non-equilibrium structures, as will be described below, provided that both peptide and lipid processes are accounted for.
A more general model for peptide-lipid interactions
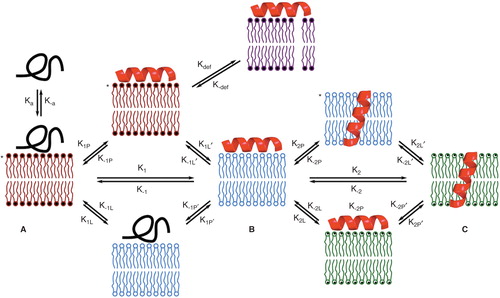
In order to account for peptide and protein association with membranes in a lipid-inclusive manner, a more general model is presented below (). The key to this model is that any change in the nature of the interaction between a peptide and a membrane, whether this involves a structural change in the peptide, peptide reorientation within the membrane, or peptide oligomerization, perturbs the bilayer structure and is therefore accompanied by a corresponding process of lipid relaxation towards an intermediate equilibrium state. Initial association with the membrane produces a state (A) in which neither the peptide nor the lipid component are at equilibrium. The membrane in this state is likely to be the highly prone to defects as it is the furthest from equilibrium. The initial membrane-bound state then relaxes to an intermediate membrane bound form (B). The equilibrium between states A and B can be considered as a direct process (with forward rate constant k1) or as microscopic equilibria for lipid and peptide relaxation with forward rate constants of k1L/k1L, and k1P/k1P' respectively. The intermediate state (B) then undergoes further transformation, such as peptide realignment, with similar lipid relaxation to give the next intermediate state (C) and so on. All states are potential precursors for thermally-induced transient defects, but the states in which the membrane component is in a more disordered (non-relaxed) state (indicated by asterisks in ) will lead to enhanced defect formation. This model is imperfect, particularly as it does not allow for co-operative interactions between peptide and lipid, and parallel pathways for the formation of peptide states, but it is nevertheless useful for addressing the outcomes of a number of peptide-membrane binding experiments.
Figure 4. A model for peptide-lipid interactions that incorporates the kinetics of both peptide and lipid-based transformations. Forward processes involving peptides and lipids have ‘P’ and ‘L’ subscripts respectively. Intermediate A is formed immediately following peptide binding. As shown here, the unfolded peptide initially binds, as occurs in many (but not all) cases. Adoption of peptide 2° structure leads to intermediate B following lipid relaxation. Intermediate B in many systems corresponds to a bilayer of reduced thickness following thinning. Intermediate C could correspond to the system following peptide insertion (as shown), but in a general scheme C would correspond to any system that involves a peptide rearrangement from B, followed by lipid relaxation. Processes such as defect formation (represented by kdef) will be favored in those intermediates (indicated by asterisks) in which the lipid component of the system is not at equilibrium. This Figure is reproduced in color in Molecular Membrane Biology online.
Initial association with the membrane
Fluorescence methods have been widely used to monitor the association of peptides with membranes. Recently, total internal reflection fluorescence (TIRF) imaging has been used to monitor the binding and unbinding of single glucagon-like peptide-1 molecules from a supported bilayer surface, leading to respective values for ka and k–a of 1.0 × 104 M-1 s-1 and 8.2 × 10-1 s-1 (Fox et al. Citation2009). An additional feature of this method is the ability to track individual peptides on the bilayer surface, giving access not only to the residence time of individual peptides (which reflects the bound state of the peptide in the membrane), but also lateral diffusion coefficients.
The intrinsic fluorescence of native tryptophan residues in the sequence of a protein provides a convenient tool for monitoring both initial binding to the membrane, and subsequent equilibria following binding. In general, initial peptide binding to the membrane is accompanied by increases in Trp fluorescence intensity and a blue shift in the emission maximum (Ladokhin and White Citation2004). For some proteins however, unexpected changes in Trp fluorescence can reveal details not accessible by other means, such as partial protein unfolding during the fast initial binding step, exemplified by red shifted emission maxima and increased fluorescence intensities following the binding of chicken liver bile acid-binding protein to negatively-charged membranes (Galassi et al. Citation2009). The same article additionally demonstrates that a single Trp residue is sufficient for experiments to monitor either intrinsic Trp fluorescence intensity, Trp as a FRET donor to NBD, or quenching of Trp fluorescence by acrylamide. The suitability of Trp as a FRET donor has also been exploited in stopped flow experiments to monitor the association of an analog of the antimicrobial peptide magainin 2 with membranes composed of POPC/POPG. This yielded a value for ka of 7.8 × 105 M-1 s-1 and a value for k–a of 88 s-1 for POPC/POPG (4:1), with ka increasing with increasing PG composition of the membrane, and k–a decreasing exponentially (Gregory et al. Citation2009).
Two methods are particularly useful for measuring the rate at which peptides adsorb and desorb from the membrane surface: surface plasmon resonance (SPR) (Mozsolits and Aguilar Citation2002, Papo and Shai Citation2003, Kamimori et al. Citation2005, El Amri et al. Citation2006) and analysis using a quartz crystal microbalance (QCM) (Wacklin Citation2011). Although these measure only the total quantity of peptide associated with the membrane, their temporal resolution is such that ka and k–a can be measured accurately, allowing the association constant to be determined as ka/k–a. For example the kinetics of binding of the protein annexin A1 to a supported bilayer with POPC/POPS (4:1) as the distal (solution-facing) leaflet were determined using a QCM, which enabled values for ka = 53 × 103 M-1 s-1 and k–a = 7 × 10-3 s-1 to be determined in the presence of 0.1 mM Ca2+ (Kastl et al. Citation2002). The inclusion of cholesterol in this system did not significantly alter the association and dissociation constants. Related experiments with annexin A2t yielded data of a similar magnitude (ka = 34 × 103 M-1 s-1 and k–a = 1.4 × 10-3 s-1) (Ross et al. Citation2003). The rate of membrane association of the bovine seminal plasma protein PDC-109 as a function of temperature and the lipid composition of supported bilayers containing 20 wt% cholesterol on a gold surface were investigated by SPR. This method gave access to both ka and k–a values. Typical values for ka at 20°C were 5.7 × 105 M-1 s-1, 1.2 × 102 M-1 s-1 and 5.3 × 102 M-1 s-1 for surface layers composed of DMPC, DMPA and DMPG respectively (Thomas et al. Citation2003). Typical respective values for k–a at 20°C were 2.7 × 10-2 s-1, 11.0 × 10-2 s-1 and 9.0 × 10-2 s-1 for the same surface layers. The kinetics of membrane association of the C2 domain of protein kinase Cα (PKCα) were found to more complex than a simple 1:1 model by SPR, but nevertheless it was apparent that the half-life for equilibration of binding was of the order of 50 s for surface layers containing PS or PI. A higher overall affinity for surface layers containing PI was reflected in a slower rate of desorbtion (k–a) in this case (Manna et al. Citation2008). SPR has equally been applied to follow the kinetics of peptide binding (Kamimori et al. Citation2005, Zhang et al. Citation2000).
A general feature of the kinetics described above is that ka is generally slower than the diffusion-controlled rate for reactions (∼1010 M-1 s-1) (Isaacs Citation1995) by several orders of magnitude, which implies that there is a free energy barrier to binding (Fox et al. Citation2009). In the case of the SPR experiments with PDC-109 (Thomas et al. Citation2003) described above, the differences in ka values were largely accounted for by entropic changes in the transition state for binding, with a positive (favourable) ▵S‡ for DMPC and a negative ▵S‡ for the other lipids.
Marker release experiments
A key question in all marker release experiments is the underlying mechanism by which membrane permeation occurs. This can be expressed in terms of whether a peptide-stabilized ensemble is formed, as in the toroidal pore, barrel stave, or carpet mechanisms, or whether permeation is a consequence of changes in bilayer stability that lead to increases in the occurrence and lifetime of transient thermally-induced defects. A fundamental issue is whether marker release kinetics are reporting peptide association with the membrane (ka) or the rate of processes that occur following association, including peptide reorganization (k1P, k1P') and lipid relaxation (k1L, k1L') or a combination of both (k1). A key requirement is identification of the rate determining step for marker release, which may be any of these processes, as well as the rate of marker diffusion along the permeation pathway (kefflux). The distinction between stable protein ensembles and efflux mediated by transient non-equilibrium structures can be made by assessing whether marker release is graded or all-or-none using fluorescence-based approaches (Ladokhin et al. Citation1997, Pokorny and Almeida Citation2004). Recently the kinetics of marker release have been used to probe the poration mechanism, with the data modeled as an initial peptide binding step followed by a transformation to a permeative state (Gregory et al. Citation2008, Citation2009). The kinetics of peptide association and dissociation were determined independently of marker release, and the rate of marker permeation (kefflux) was accounted for, bringing this method close to model in without direct consideration of membrane equilibration. No assumptions were made concerning the nature of the permeative state, although a model that included peptide oligomerization did not fit the data well. Using this approach, cecropin A could be shown to operate with an all-or-none mechanism in POPC/POPG membranes, whereas analogs of magainin 2 changed from an all-or-none mechanism to a graded mechanism as the PG content of POPC/POPG membranes fell below ∼ 30 mol%. It is notable that the kinetics of marker release in these experiments were fast, with t1/2 typically <1 min for cecropin A with membranes composed of POPC/POPG (1:1), and ∼6 min for POPC/POPG (4:1). Equivalent values for magainin 2 were t1/2 ∼ 4 min and ∼ 20 min, respectively, in the same lipid systems. A rigorous study has addressed marker release induced by the peptide pardaxin from liposomes composed of either DOPC or DOPC/DOPG (4:1) containing calcein as the marker (Vad et al. Citation2010). The data demonstrate complex biphasic marker release kinetics in response to changes of pH and peptide concentration. For DOPC, marker release occurred more rapidly with increasing pH and reached a saturating value of t1/2 = 0.02 min at pH 10 for a P/L of ≈ 0.0002 for the fast phase, and t1/2 = 0.2 min at pH 10 for a P/L of ≈ 0.0005 for the slow phase. At the same P/L at pH 5.5, the t1/2 for the slow phase increases to several minutes. For DOPC/DOPG, the rates of marker release were fastest at pH 5.5, with values for t1/2 of 0.01 min (P/L ≈ 0.005) and 0.07 min (P/L ≈ 0.01) being typical for the fast and slow phases, respectively. Further experiments were conducted by 13C NMR, which indicated that the peptide inserts into a transmembrane arrangement in DOPC liposomes, but remains peripherally bound in membranes containing DOPG. The marker efflux kinetics for pardaxin can be accounted for by complementarity between the net charge of the peptide and the membrane at specific pH values, and differences in peptide rearrangement that occur following initial membrane association. The examples above illustrate that the processes which lead to marker efflux are inherently rapid for both defect-mediated poration and peptide-mediated poration where the peptide remains bound peripherally, which is in accord with the in-plane diffusion coefficients (DL) for lipids and proteins, which are the same order of magnitude in model systems where both are addressed (although peptides and proteins diffuse more slowly than lipids in accordance with their higher molecular weight). The kinetics of peptide insertion to transmembrane configurations are more varied and are discussed in more detail in the next section.
In many cases, marker release is accompanied by translocation of lipids between leaflets, with both processes occurring on a similar timescale following peptide administration (Arbuzova and Schwarz Citation1999, Pokorny and Almeida Citation2004, Fuertes et al. Citation2011, Kotova et al. Citation2011). The kinetics data therefore suggest that rapid membrane poration and lipid translocation share a common mechanism. Mechanisms that involve increased lipid disorder, whether directly as a response to local perturbation following peptide binding (Fernandez et al. Citation2011), or as a consequence of membrane thinning, are good candidates. The formation of transient defects that enable significant water ingress into the hydrophobic interior of the bilayer will benefit both lipid translocation and membrane permeation in the same manner by reducing the requirements for desolvation.
Changes in peptide-membrane systems occurring over longer time scales
In many of the cases discussed above, initial rapid change of the experimental parameter being monitored is followed by one or more slower phases. Slow kinetic processes are typically observed in situations that involve peptide insertion to transmembrane arrangements, such as the case for pardaxin in DOPC membranes (Vad et al. Citation2010). In terms of the model in , this corresponds to kP2 and kP2', as well as the corresponding lipid relaxation processes (kL2 and kL2). Transmembrane insertion is inherently sensitive to complementarity between the net charges of the peptide and the membrane, which is apparent in the case of pardaxin association with neutral DOPC described above, where decreasing the pH increases the net charge on the peptide, with concomitant decreases in the rate of peptide insertion and marker efflux. Insertion of the model peptide TMX-3, when monitored by intrinsic Trp fluorescence, similarly exhibits significantly slower insertion kinetics at pH 5 (>60 min) when compared with pH 6.3 (∼1 min) (Ladokhin and White Citation2004).
Direct measurements of peptide insertion that follow changes in peptide spectral properties, such as intrinsic Trp fluorescence in the example above, are a robust means of quantifying slow kinetic processes. Linear dichroism (LD) spectroscopy and oriented circular dichroism spectroscopy (OCD) similarly provide direct measurements of peptide behavior in membranes, both yielding data that reveal the orientation of absorbing chromophores. When applied to studying the membrane association of peptides, the predominant chromophores arise from the peptide backbone and aromatic side chains, as lipids have extremely weak chromophores. OCD is closely related to conventional CD spectroscopy, with the difference being that the membranes are aligned with respect to the experimental frame (Wu et al. Citation1990, Yang et al. Citation2001, Chen et al. Citation2003, Lee et al. Citation2004, Citation2005, Huang Citation2006, Bürck et al. Citation2008, Qian et al. Citation2008, Cheng et al. Citation2010). LD spectroscopy measures the differential absorbance by the sample of linear and parallel polarized light (Rodger et al. Citation2002, Castanho et al. Citation2003, Caesar et al. Citation2006, Esbjörner et al. Citation2007, Svensson et al. Citation2011). An aligned sample is an absolute requirement for LD: non-aligned samples do not yield an LD spectrum. Alignment is usually achieved using bilayers adsorbed in glass plates, or by shear flow (Dafforn et al. Citation2004, Marrington et al. Citation2005). Changes in the alignment of a peptide with regard to the membrane can be monitored by both LD and OCD in real time. LD spectroscopy has the additional advantage that peptides not associated with the membrane are not aligned and therefore do not contribute to the observed signal. In these experiments, only changes in peptide orientation are monitored; as a consequence the data are interpreted in terms of k1/k-1 and k2/k-2 (). Studies by OCD and LD can detect the presence of multiple peptide equilibria and have revealed that models that involve equilibria between peripheral and membrane inserted states are accurate for many systems (Ennaceur et al. Citation2009, Svensson et al. Citation2011). It is apparent from these experiments that complete equilibration of the peptide-lipid system is only achieved after significant periods of time. For example, association of the peptide melittin with DMPC membranes yielded complex behavior when studied by LD, requiring >7 h for equilibration at 25°C and ∼ 10 min at 37°C (CitationDamianoglou et al. 2010). Similar periods of hours were required for complete equilibration in POPC/DOPG (4:1) membranes (Svensson et al. Citation2011). Model peptides have similarly yielded slow rates for k2, attributed to membrane insertion on the basis of the LD data, with a t1/2 of 75–100 min (Ennaceur et al. Citation2009).
Esbjörner et al. (Citation2007) studied the association of the HA2 fusion peptide from influenza virus hemagglutinin with synthetic membranes composed of POPC or POPC/POPG (4:1) using a range of methods, including LD and intrinsic fluorescence. The latter approach further enabled the fast association with the membrane (ka) to be resolved, with a t1/2 of < 2 s. Interestingly, marker release experiments revealed membrane poration only during this fast kinetic phase, with marker release more prominent from POPC vesicles. A second, slower phase was detectable by both intrinsic fluorescence and LD, with a t1/2 > 1 h. A key feature of this work was the use of membrane-embedded retinoic acid as an LD marker for membrane order. This enabled the kinetics of membrane relaxation to be followed in tandem with changes in peptide orientation. Two fundamental observations resulted: Firstly, the extent of marker release in the fast phase correlated with the degree of membrane disorder in this phase; secondly equilibration of the LD signal from retinoic acid required periods in excess of 1 h, indicating that membrane relaxation in this system is slow and potentially coupled to protein reorientation. NMR relaxation experiments have demonstrated that even in the equilibrium state, there can still be significant peptide rocking motions of hemagglutinin with respect to the membrane (Lorieau et al. Citation2011), which acts as a salient reminder that spectroscopic methods such as fluorescence and LD measure the average properties of the species in question.
It has recently become evident that even in simple model peptide-lipid systems, the lipid cannot be considered as an inert medium in which peptide binding and reorientation occurs, with acyl transfer from the lipid to the peptide melittin from POPC membranes, in the absence of enzyme catalysis, detectable as a background reaction with a t1/2 of ≈ 16 days in phosphate buffered saline (Pridmore et al. Citation2011). The same reaction has a t1/2 of ≈ 24 h in bicarbonate buffer at pH 7.4, with acylation products detectable after 4 h (Dods et al. Citation2012). Acylation occurs primarily at the N-terminal amino group of melittin, as well as the amino groups of lysine side chains, most notably the lysine closest to the C-terminus of the peptide. Traces of doubly-acylated peptide, alongside putative acyl transfer to the side chain of a serine, could also be detected. The extent to which this reactivity is typical for membrane-active peptides and proteins is yet to be established. Anomalous behavior observed with other peptides, such as TMX-3, which displayed irreversible membrane binding (Ladokhin and White Citation2004), may also be the consequence of peptide acylation by lipids, although this is currently unproven.
In vivo studies that address both early and late stages of binding
Studies on the association of peptides with cell membranes in vivo generally reveal that fast processes such as membrane depolarization occur on a timescale that is slower than the ka in model systems, but nevertheless still sufficiently rapid that t1/2 values are < 10 min. 125I-labeled streptolysin O exhibited biphasic binding kinetics with red blood cell membranes, with a fast initial binding (t1/2 ∼ 70 s) at 37°C followed by a slower phase with a timescale of (t1/2 ∼ 10 min) during which streptolysin O oligomers assembled (Palmer et al. Citation1995). The binding of a number of peptides to Staphylococcus aureus membranes was assayed by addressing the rate of membrane depolarization and survival rate following peptide administration. In general, depolarization occurred with a t1/2 of 2–3 min, with cell death requiring longer time frames (tens of minutes) (Friedrich et al. Citation2000). A similar t1/2 for membrane depolarization was observed following the administration of aurein analogs to S. aureus C622 (Cheng et al. Citation2010). Addition of melittin to sheep lymphocytes loaded with calcein yielded distinct biphasic marker release kinetics, with a fast initial release of marker (t1/2 ∼ 1 min) and a much slower second phase (t1/2 ∼ 1 h) (Su et al. Citation2001).
Recent improvements in atomic force microscopy have enabled individual images to be obtained within a time span of 10–15 s or less, making high-speed AFM a viable tool for studying peptide- and protein-membrane binding. This has enabled the effects of administration of the antimicrobial peptide CM15 to Escherichia coli to be monitored in real time (Fantner et al. Citation2010). Noticeable roughening of the surface of the bacterium was detectable within 2 min of administration of the peptide, consistent with a rapid initial adsorbtion of the peptide (t1/2 = 52 ± 16 s) and a t1/2 for bulk killing of 4.6 min.
Summary of peptide binding kinetics
From the arguments presented above, some general features of the association of peptides and proteins with lipid membranes can be described. The initial rate of association (ka) is fast (t1/2 of milliseconds to seconds) and 1 to 5 orders of magnitude slower than the diffusion of water. Peptide dissociation from the membrane (k–a) is generally 3 to 6 orders of magnitude slower than association, depending on the strength of the association with the membrane. Following peptide association, both initial structural changes to the peptide, and membrane reorganization are fast (t1/2 of milliseconds to seconds), and assembly to permeative states also fast (t1/2 of seconds). Rates of subsequent processes, particularly membrane reorientation (i.e., insertion to transmembrane conformations) yield much greater variations in rate profiles, with t1/2 values ranging from seconds to hours.
Conclusions
The data obtained from model systems have demonstrated the fundamental properties of lateral, transverse and rotational diffusion in membranes. Nevertheless, a number of recent observations have fuelled debates that have implications for our understanding of both model and biogenic systems. One of the foremost issues concerns the structure of SLBs. A better understanding of the structure of SLBs at the molecular level will be central to comprehending the atypical properties exhibited by some SLBs, such as fast transverse diffusion and electrostatically-induced asymmetry. Biogenic membranes exhibit more diverse behavior than their model counterparts, largely as a consequence of a high protein content, more complex lipid composition, and interactions with the cytoskeleton. Whilst molecular diffusion in biogenic membranes is generally slower than in model systems, anomalous diffusion in biological systems is widely reported, manifested by localized variations in diffusion rates. Understanding why phenomena such as anomalous or confined diffusion are specific to certain membranes or induced by under specific conditions will lead to a better understanding of the kinetics of membrane processes such as receptor clustering and membrane remodelling. The role of cholesterol in moderating membrane properties continues to provoke much argument. This is reflected both by the large diversity of reported transverse diffusion rates and the inability to observe the macroscopic phase separation in vivo that is seen in vitro. Improvements to the methods available for studying the lateral diffusion of membrane components in biological membranes, such as FCS-STED and NMR, will enhance our understanding of how cholesterol interactions with specific lipids classes lead to lateral asymmetry and the formation of rafts, as well as the more general role of cholesterol in moderating membrane fluidity.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Anglin TC, Conboy JC. 2009. Kinetics and thermodynamics of flip-flop in binary phospholipid membranes measured by sum-frequency vibrational spectroscopy. Biochemistry 48:10220–10234.
- Anglin TC, Brown KL, Conboy JC. 2009. Phospholipid flip-flop modulated by transmembrane peptides WALP and melittin. J Struct Biol 168:37–52.
- Anglin TC, Cooper MP, Li H, Chandler K, Conboy JC. 2010. Free energy and entropy of activation for phospholipid flip-flop in planar supported lipid bilayers. J Phys Chem B 114:1903–1914.
- Anglin TC, Liu J, Conboy JC. 2007. Facile lipid flip-flop in a phospholipid bilayer induced by gramicidin A measured by sum-frequency vibrational spectroscopy. Biophys J 92:L01–L03.
- Apellániz B, Nieva JL, Schwille P, García-Sáez AJ. 2010. All-or-none versus graded: Single-vesicle analysis reveals lipid composition effects on membrane permeabilization. Biophys J 99:3619–3628.
- Arbuzova A, Schwarz G. 1999. Pore-forming action of mastoparan peptides on liposomes: A quantitative analysis. Biochim Biophys Acta 1420:139–152.
- Ariola FS, Li Z, Cornejo C, Bittman R, Heikal AA. 2009. Membrane fluidity and lipid order in ternary giant unilamellar vesicles using a new Bodipy-cholesterol derivative. Biophys J 96:2696–2708.
- Ayuyan AG, Cohen FS. 2008. Raft composition at physiological temperature and pH in the absence of detergents. Biophys J 94:2654–2666.
- Baier CJ, Gallegos CE, Levi V, Barrantes FJ. 2010. Cholesterol modulation of nicotinic acetylcholine receptor surface mobility. Eur Biophys J 39:213–227.
- Baumgart T, Hunt G, Farkas ER, Webb WW, Feigenson GW. 2007. Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim Biophys Acta 1768:2182–2194.
- Bechinger B, Lohner K. 2006. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta 1758:1529–1539.
- Berkovich R, Wolfenson H, Eisenberg S, Ehrlich M, Weiss M, Klafter J, 2011. Accurate quantification of diffusion and binding kinetics of non-integral membrane proteins by FRAP. Traffic 12:1648–1657.
- Biju V, Itoh T, Ishikawa M. 2010. Delivering quantum dots to cells: Bioconjugated quantum dots for targeted and nonspecific extracellular and intracellular imaging. Chem Soc Rev 39:3031–3056.
- Bocchinfuso G, Bobone S, Mazzuca C, Palleschi A, Stella L. 2011. Fluorescence spectroscopy and molecular dynamics simulations in studies on the mechanism of membrane destabilization by antimicrobial peptides. Cell Mol Life Sci 68:2281–2301.
- Boon JM, Smith BD. 2002. Chemical control of phospholipid distribution across bilayer membranes. Med Res Rev 22:251–281.
- Brown DA. 2006. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21:430–439.
- Brown KL, Conboy JC. 2011. Electrostatic induction of lipid asymmetry. J Am Chem Soc 133:8794–8797.
- Bruckner RJ, Mansy SS, Ricardo A, Mahadevan L, Szostak JW. 2009. Flip-flop-induced relaxation of bending energy: Implications for membrane remodeling. Biophys J 97:3113–3122.
- Bürck J, Roth S, Wadhwani P, Afonin S, Kanithasen N, Strandberg E, 2008. Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys J 95:3872–3881.
- Busch S, Smuda C, Pardo LC, Unruh T. 2010. Molecular mechanism of long-range diffusion in phospholipid membranes studied by quasielastic neutron scattering. J Am Chem Soc 132:3232–3233.
- Caesar CEB, Esbjörner EK, Lincoln P, Nordén B. 2006. Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: Correlations of structure to cellular uptake. Biochemistry 45:7682–7692.
- Calvo-Muñoz EM, Selvan ME, Xiong R, Ojha M, Keffer DJ, Nicholson DM, 2011. Applications of a general random-walk theory for confined diffusion. Phys Rev E 83:011120.
- Castanho M, Lopes S, Fernandes M. 2003. Using UV-Vis. linear dichroism to study the orientation of molecular probes and biomolecules in lipidic membranes. Spectrosc Int J 17:377–398.
- Castellana ET, Cremer PS. 2006. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf Sci Rep 61:429–444.
- Casuso I, Rico F, Scheuring S. 2011. High-speed atomic force microscopy: Structure and dynamics of single proteins. Curr Opin Chem Biol 15:704–709.
- Chakraborty A, Kim S, Snyder SH. 2011. Inositol pyrophosphates as mammalian cell signals. Sci Signal 4; re1.
- Chen F-Y, Lee M-T, Huang HW. 2003. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys J 84:3751–3758.
- Cheng JTJ, Hale JD, Kindrachuk J, Jessen H, Elliott M, Hancock REW, 2010. Importance of residue 13 and the C-terminus for the structure and activity of the antimicrobial peptide aurein 2.2. Biophys J 99:2926–2935.
- Cherry RJ, Godfrey RE. 1981. Anisotropic rotation of bacteriorhodopsin in lipid membranes. Comparison of theory with experiment. Biophys J 36:257–276.
- Cho N-J, Cho S-J, Hardesty JO, Glenn JS, Frank CW. 2007. Creation of lipid partitions by deposition of amphipathic viral peptides. Langmuir 23:10855–10863.
- Cornell BA, Separovic F, Baldassi AJ, Smith R. 1988. Conformation and orientation of gramicidin A in oriented phospholipid bilayers measured by solid state carbon-13 NMR. Biophys J 53:67–76.
- Crane JM, Kiessling V, Tamm LK. 2005. Measuring lipid asymmetry in planar supported bilayers by fluorescence interference contrast microscopy. Langmuir 21:1377–1388.
- Crane JM, Verkman AS. 2008. Long-range nonanomalous diffusion of quantum dot-labeled aquaporin-1 water channels in the cell plasma membrane. Biophys J 94:702–713.
- Crane JM, Van Hoek AN, Skach WR, Verkman AS. 2008. Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Mol Biol Cell 19:3369–3378.
- Crane JM, Tajima M, Verkman AS. 2010. Live-cell imaging of aquaporin-4 diffusion and interactions in orthogonal arrays of particles. Neuroscience 1684:892–902.
- Czolkos I, Jesorka A, Orwar O. 2011. Molecular phospholipid films on solid supports. Soft Matter 7:4562–4576.
- Dafforn TR, Rajendra J, Halsall DJ, Serpell LC, Rodger A. 2004. Protein fiber linear dichroism for structure determination and kinetics in a low-volume, low-wavelength couette flow cell. Biophys J 86:404–410.
- Daleke DL. 2003. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res 44:233–242.
- Damianoglou A, Rodger A, Pridmore C, Dafforn TR, Mosely JA. Sanderson, JM, Hicks MR. 2010. The synergistic action of melittin and phospholipase A2 with lipid membranes: Development of linear dichroism for membrane-insertion kinetics. Protein Pept Lett 17:1351–1362.
- Day CA, Kenworthy AK. 2009. Tracking microdomain dynamics in cell membranes. Biochim Biophys Acta 1788:245–253.
- De Angelis AA, Grant CV, Baxter MK, McGavin JA, Opella SJ, Cotten ML. 2011. Amphipathic antimicrobial piscidin in magnetically aligned lipid bilayers. Biophys J 101:1086–1094.
- De Kruijff B, Van Zoelen EJ. 1978. Effect of the phase transition on the transbilayer movement of dimyristoyl phosphatidylcholine in unilamellar vesicles. Biochim Biophys Acta 511:105–115.
- Delint-Ramirez I, Willoughby D, Hammond GVR, Ayling LJ, Cooper DMF. 2011. Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J Biol Chem 286:32962–32975.
- Devaux PF, Zachowski A. 1994. Maintenance and consequences of membrane phospholipid asymmetry. Chem Phys Lipids 73:107–120.
- Devaux PF, Herrmann A. 2012. Transmembrane dynamics of lipids. Hoboken, NJ: Wiley.
- Dix JA, Verkman AS. 2008. Crowding effects on diffusion in solutions and cells. Ann Rev Biophys 37:247–263.
- Dods RH, Mosely JA, Sanderson JM. 2012. The innate reactivity of a membrane associated peptide towards lipids: Acyl transfer to melittin without enzyme catalysis. Org Biomol Chem in press; doi: 10.1039/c2ob07113d.
- Domanov YA, Aimon S, Toombes GES, Renner M, Quemeneur F, Triller A, 2011. Mobility in geometrically confined membranes. Proc Natl Acad Sci USA 108:12605–12610.
- Duggan J, Jamal G, Tilley M, Davis B, Mckenzie G, Vere K, 2008. Functional imaging of microdomains in cell membranes. Eur Biophys J 37:1279–1289.
- Dupuy AD, Engelman DM. 2008. Protein area occupancy at the center of the red blood cell membrane. Proc Natl Acad Sci USA 105:2848–2852.
- Eckford PDW, Sharom FJ. 2010. The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem J 429:195–203.
- Eisenthal KB. 2006. Second harmonic spectroscopy of aqueous nano- and microparticle interfaces. Chem Rev 106:1462–1477.
- El Amri C, Lacombe C, Zimmerman K, Ladram A, Amiche M, Nicolas P, 2006. The plasticins: Membrane adsorption, lipid disorders, and biological activity. Biochemistry 45:14285–14297.
- Ennaceur SM, Hicks MR, Pridmore CJ, Dafforn TR, Rodger A, Sanderson JM. 2009. Peptide adsorption to lipid bilayers: Slow processes revealed by linear dichroism spectroscopy. Biophys J 96:1399–1407.
- Epand RM, Epand RF. 2009. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim Biophys Acta 1788:289–294.
- Esbjörner EK, Oglecka K, Lincoln P, Gräslund A, Nordén B. 2007. Membrane binding of pH-sensitive influenza fusion peptides. Positioning, configuration, and induced leakage in a lipid vesicle model. Biochemistry 46:13490–13504.
- Etemadi AH. 1980. Membrane asymmetry. A survey and critical appraisal of the methodology. II. Methods for assessing the unequal distribution of lipids. Biochim Biophys Acta 604:423–475.
- Falck E, Róg T, Karttunen M, Vattulainen I. 2008. Lateral diffusion in lipid membranes through collective flows. J Am Chem Soc 130:44–45.
- Fantner GE, Barbero RJ, Gray DS, Belcher AM. 2010. Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat Nanotechnol 5:280–285.
- Fattal E, Nir S, Parente RA, Szoka FC. 1994. Pore-forming peptides induce rapid phospholipid flip-flop in membranes. Biochemistry 33:6721–6731.
- Fernandez DI, Sani M-A, Separovic F. 2011. Interactions of the antimicrobial peptide Maculatin 1.1 and analogues with phospholipid bilayers. Aust J Chem 64:798–805.
- Filippov A, Orädd G, Lindblom G. 2003. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys J 84:3079–3086.
- Fooksman DR, Edidin M, Barisas BG. 2007. Measuring rotational diffusion of MHC class I on live cells by polarized FPR. Biophys Chem 130:10–16.
- Fox CB, Wayment JR, Myers GA, Endicott SK, Harris JM. 2009. Single-molecule fluorescence imaging of peptide binding to supported lipid bilayers. Anal Chem 81:5130–5138.
- Frasch SC, Henson PM, Nagaosa K, Fessler MB, Borregaard N, Bratton DL. 2004. Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated Met-Leu-Phe-stimulated neutrophils. J Biol Chem 279:17625–17633.
- Frick M, Schmidt K, Nichols BJ. 2007. Modulation of lateral diffusion in the plasma membrane by protein density. Curr Biol 17:462–467.
- Friedrich CL, Moyles D, Beveridge TJ, Hancock RE. 2000. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob Agents Chemother 44:2086–2092.
- Fuertes G, García-Sáez AJ, Esteban-Martín S, Giménez D, Sánchez-Muñoz OL, Schwille P, 2010. Pores formed by baxα5 relax to a smaller size and keep at equilibrium. Biophys J 99:2917–2925.
- Fuertes G, Giménez D, Esteban-Martín S, Sánchez-Muñoz OL, Salgado J. 2011. A lipocentric view of peptide-induced pores. Eur Biophys J 40:399–415.
- Galassi V, Nolan V, Villarreal MA, Perduca M, Monaco HL, Montich GG. 2009. Kinetics of lipid-membrane binding and conformational change of L-BABP. Biochem Bioph Res Co 382:771–775.
- Gambin Y, Reffay M, Sierecki E, Homblé F, Hodges RS, Gov NS, 2010. Variation of the lateral mobility of transmembrane peptides with hydrophobic mismatch. J Phys Chem B 114:3559–3566.
- Garg S, Porcar L, Woodka AC, Butler PD, Perez-Salas U. 2011. Noninvasive neutron scattering measurements reveal slower cholesterol transport in model lipid membranes. Biophys J 101:370–377.
- Ge M, Freed JH. 2011. Two conserved residues are important for inducing highly ordered membrane domains by the transmembrane domain of influenza hemagglutinin. Biophys J 100:90–97.
- Golebiewska U, Kay JG, Masters T, Grinstein S, Im W, Pastor RW, 2011. Evidence for a fence that impedes the diffusion of phosphatidylinositol 4,5-bisphosphate out of the forming phagosomes of macrophages. Mol Biol Cell 22:3498–3507.
- Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. 2008. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell 19:1663–1669.
- Gregory SM, Cavenaugh A, Journigan V, Pokorny A, Almeida PFF. 2008. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys J 94:1667–1680.
- Gregory SM, Pokorny A, Almeida PFF. 2009. Magainin 2 revisited: A test of the quantitative model for the all-or-none permeabilization of phospholipid vesicles. Biophys J 96:116–131.
- Gurtovenko AA, Vattulainen I. 2005. Pore formation coupled to ion transport through lipid membranes as induced by transmembrane ionic charge imbalance: Atomistic molecular dynamics study, J Am Chem Soc. 127:17570–17571.
- Gurtovenko AA, Vattulainen I. 2007a. Molecular mechanism for lipid flip-flops. J Phys Chem B 111:13554–13559.
- Gurtovenko AA, Vattulainen I. 2007b. Ion leakage through transient water pores in protein-free lipid membranes driven by transmembrane ionic charge imbalance. Biophys J 92:1878–1890.
- Gurtovenko AA, Anwar J, Vattulainen I. 2010. Defect-mediated trafficking across cell membranes: Insights from in silico modeling. Chem Rev 110:6077–6103.
- Haggie PM, Kim JK, Lukacs GL, Verkman AS. 2006. Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol Biol Cell 17:4937–4945.
- Hancock JF. 2006. Lipid rafts: Contentious only from simplistic standpoints. Nat Rev Mol Cell Biol 7:456–462.
- Hennig M, Neumann J, Wixforth A, Rädler JO, Schneider MF. 2009. Dynamic patterns in a supported lipid bilayer driven by standing surface acoustic waves. Lab Chip 9:3050–3053.
- Honerkamp-Smith AR, Veatch SL, Keller SL. 2009. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochim Biophys Acta 1788:53–63.
- Horng ML, Gardecki JA, Papazyan A, Maroncelli M. 1995. Subpicosecond measurements of polar solvation dynamics: Coumarin-153 revisited. J Phys Chem 99:17311–17337.
- Horst R, Horwich AL, Wüthrich K. 2011. Translational diffusion of macromolecular assemblies measured using transverse-relaxation-optimized pulsed field gradient NMR. J Am Chem Soc 133:16354–16357.
- Horton MR, Hoefling F, Raedler JO, Franosch T. 2010. Development of anomalous diffusion among crowding proteins. Soft Matter 6:2648–2656.
- Huang HW. 2006. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim Biophys Acta 1758:1292–1302.
- Huang HW, Chen F-Y, Lee M-T. 2004. Molecular mechanism of peptide-induced pores in membranes. Phys Rev Lett. 92:198304.
- Hurley JH. 2006. Membrane binding domains. Biochim Biophys Acta 1761:805–811.
- Hurley JH, Meyer T. 2001. Subcellular targeting by membrane lipids. Curr Opin Cell Biol 13:146–152.
- Isaacs N. 1995. Physical organic chemistry. Harlow, UK: Longman.
- Jacobson K, Mouritsen OG, Anderson RGW. 2007. Lipid rafts: At a crossroad between cell biology and physics. Nat Cell Biol 9:7–14.
- Johnson S, Bayerl T, McDermott D, Adam G, Rennie A, Thomas R, 1991. Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys J 59:289–294.
- Kahya N, Schwille P. 2006. How phospholipid-cholesterol interactions modulate lipid lateral diffusion, as revealed by fluorescence correlation spectroscopy. J Fluoresc 16:671–678.
- Kahya N, Scherfeld D, Bacia K, Poolman B, Schwille P. 2003. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J Biol Chem 278:28109–28115.
- Kamimori H, Blazyk J, Aguilar M-I. 2005. Lipid membrane-binding properties of tryptophan analogues of linear amphipathic beta-sheet cationic antimicrobial peptides using surface plasmon resonance. Biol Pharm Bull 28:148–150.
- Kastl K, Ross M, Gerke V, Steinem C. 2002. Kinetics and thermodynamics of annexin A1 binding to solid-supported membranes: A QCM study. Biochemistry 41:10087–10094.
- Kaya Aİ, Uğur Ö, Altuntaş O, Sayar K, Onaran HO. 2011. Long and short distance movements of β2-adrenoceptor in cell membrane assessed by photoconvertible fluorescent protein dendra2-β2-adrenoceptor fusion. Biochim Biophys Acta 1813:1511–1524.
- Kiessling V, Crane JM, Tamm LK. 2006. Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys J 91:3313–3326.
- Kol MA, de Kroon AIPM, Rijkers DTS, Killian JA, de Kruijff B. 2001. Membrane-spanning peptides induce phospholipid flop: A model for phospholipid translocation across the inner membrane of E. coli. Biochemistry 40:10500–10506.
- Kol MA, van Laak ANC, Rijkers DTS, Killian JA, de Kroon AIPM, de Kruijff B. 2003. Phospholipid flop induced by transmembrane peptides in model membranes is modulated by lipid composition. Biochemistry 42:231–237.
- Kornberg RD, McConnell HM. 1971. Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry 10:1111–1120.
- Kotova EA, Kuzevanov AV, Pashkovskaya AA, Antonenko YN. 2011. Selective permeabilization of lipid membranes by photodynamic action via formation of hydrophobic defects or pre-pores. Biochim Biophys Acta 1808:2252–2257.
- Kusumi A, Shirai YM, Koyama-Honda I, Suzuki KGN, Fujiwara TK. 2010. Hierarchical organization of the plasma membrane: Investigations by single-molecule tracking vs. fluorescence correlation spectroscopy. FEBS Lett 584:1814–1823.
- Kusumi A, Suzuki KGN, Kasai RS, Ritchie K, Fujiwara TK. 2011. Hierarchical mesoscale domain organization of the plasma membrane. Trends Biochem Sci 36:604–615.
- Kułakowska A, Jurkiewicz P, Sýkora J, Benda A, Mely Y, Hof M. 2010. Fluorescence lifetime tuning – a novel approach to study flip-flop kinetics in supported phospholipid bilayers. J Fluoresc 20:563–569.
- Ladokhin AS, White SH. 2004. Interfacial folding and membrane insertion of a designed helical peptide. Biochemistry 43:5782–5791.
- Ladokhin AS, Wimley WC, Hristova KWC, White SH. 1997. Mechanism of leakage of contents of membrane vesicles determined by fluorescence requenching. Methods Enzymol 278:474–486.
- Lagerholm BC, Weinreb GE, Jacobson K, Thompson NL. 2005. Detecting microdomains in intact cell membranes. Annu Rev Phys Chem 56:309–336.
- Langer M, Langosch D. 2011. Is lipid flippase activity of SNARE transmembrane domains required for membrane fusion. FEBS Lett 585:1021–1024.
- Lee M-T, Chen FY, Huang HW. 2004. Energetics of pore formation induced by membrane active peptides. Biochemistry 43:3590–3599.
- Lee M-T, Hung W-C, Chen F-Y, Huang HW. 2005. Many-body effect of antimicrobial peptides: On the correlation between lipid's spontaneous curvature and pore formation. Biophys J 89:4006–4016.
- Lee M-T, Hung W-C, Chen F-Y, Huang HW. 2008. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc Natl Acad Sci USA 105:5087–5092.
- Lenard J, Rothman JE. 1976. Transbilayer distribution and movement of cholesterol and phospholipid in the membrane of influenza virus. Proc Natl Acad Sci USA 73:391–395.
- Lichtenberg D, Goñi FM, Heerklotz H. 2005. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem Sci 30:430–436.
- Lindblom G, Orädd G, Filippov A. 2006. Lipid lateral diffusion in bilayers with phosphatidylcholine, sphingomyelin and cholesterol. Chem Phys Lipids 141:179–184.
- Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50.
- Liu J, Conboy JC. 2004. Direct measurement of the transbilayer movement of phospholipids by sum-frequency vibrational spectroscopy. J Am Chem Soc 126:8376–8377.
- Liu J, Conboy JC. 2005. 1,2-diacyl-phosphatidylcholine flip-flop measured directly by sum-frequency vibrational spectroscopy. Biophys J 89:2522–2532.
- Longo ML, Waring AJ, Gordon LM, Hammer DA. 1998. Area expansion and permeation of phospholipid membrane bilayers by influenza fusion peptides and melittin. Langmuir 14:2385–2395.
- Lorieau JL, Louis JM, Bax A. 2011. Whole-body rocking motion of a fusion peptide in lipid bilayers from size-dispersed 15N NMR relaxation. J Am Chem Soc 133:14184–14187.
- Loura LMS, de Almeida RFM, Silva LC, Prieto M. 2009. FRET analysis of domain formation and properties in complex membrane systems. Biochim Biophys Acta 1788:209–224.
- Ludtke S, He K, Huang H. 1995. Membrane thinning caused by magainin 2. Biochemistry 34:16764–16769.
- Macdonald PM, Soong R. 2011. Diffusion NMR and bicelle morphology. Can J Chem 89:1021–1035.
- Mainali L, Feix JB, Hyde JS, Subczynski WK. 2011. Membrane fluidity profiles as deduced by saturation-recovery EPR measurements of spin-lattice relaxation times of spin labels. J Magn Reson 212:418–425.
- Manna D, Bhardwaj N, Vora MS, Stahelin RV, Lu H, Cho W. 2008. Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKCα C2 domain. J Biol Chem 283:26047–26058.
- Marrington R, Dafforn TR, Halsall DJ, MacDonald JI, Hicks M, Rodger A. 2005. Validation of new microvolume Couette flow linear dichroism cells. The Analyst 130:1608–1616.
- Marrink SJ, de Vries AH, Tieleman DP. 2009. Lipids on the move: Simulations of membrane pores, domains, stalks and curves. Biochim Biophys Acta 1788:149–168.
- Marsh D. 2008. Protein modulation of lipids, and vice-versa, in membranes. Biochim Biophys Acta 1778:1545–1575.
- Matsuzaki K, Murase O, Fujii N, Miyajima K. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 35:11361–11368.
- Mazzuca C, Orioni B, Coletta M, Formaggio F, Toniolo C, Maulucci G, 2010. Fluctuations and the rate-limiting step of peptide-induced membrane leakage. Biophys J 99:1791–1800.
- Melo AM, Prieto M, Coutinho A. 2011. The effect of variable liposome brightness on quantifying lipid-protein interactions using fluorescence correlation spectroscopy. Biochim Biophys Acta 1808:2559–2568.
- Menon AK, Herrmann A. 2012. Phospholipid flip-flop in biogenic membranes. In: Devaux PF, Herrmann A, editors. Transmembrane dynamics of lipids. Hoboken, NJ: Wiley. 97–118.
- Metkar SS, Wang B, Catalan E, Anderluh G, Gilbert RJC, Pardo J, 2011. Perforin rapidly induces plasma membrane phospholipid flip-flop. PLoS One 6:e24286.
- Mika JT, Poolman B. 2011. Macromolecule diffusion and confinement in prokaryotic cells. Curr Opin Biotechnol 22:117–126.
- Mozsolits H, Aguilar MI. 2002. Surface plasmon resonance spectroscopy: An emerging tool for the study of peptide-membrane interactions. Biopolymers 66:3–18.
- Mueller V, Ringemann C, Honigmann A, Schwarzmann G, Medda R, Leutenegger M, 2011. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys J 101:1651–1660.
- Müller P, Herrmann A. 2002. Rapid transbilayer movement of spin-labeled steroids in human erythrocytes and in liposomes. Biophys J 82:1418–1428.
- Nakano M, Fukuda M, Kudo T, Matsuzaki N, Azuma T, Sekine K, 2009. Flip-flop of phospholipids in vesicles: Kinetic analysis with time-resolved small-angle neutron scattering. J Phys Chem B 113:6745–6748.
- Naumann CA, Prucker O, Lehmann T, Rühe J, Frank CW. 2002. The polymer-supported phospholipid bilayer: Tethering as a new approach to substrate-membrane stabilization. Biomacromolecules 3:27–35.
- Nechyporuk-Zloy V, Dieterich P, Oberleithner H, Stock C, Schwab A. 2008. Dynamics of single potassium channel proteins in the plasma membrane of migrating cells. Am J Physiol-Cell Ph 294:C1096–C1102.
- Noda I. 2010. Two-dimensional correlation spectroscopy – biannual survey 2007–2009. J Mol Struct 974:3–24.
- Orädd G, Lindblom G, Westerman P. 2002. Lateral diffusion of cholesterol and dimyristoylphosphatidylcholine in a lipid bilayer measured by pulsed field gradient NMR spectroscopy. Biophys J 83:2702–2704.
- Owen DM, Gaus K, Magee AI, Cebecauer M. 2010. Dynamic organization of lymphocyte plasma membrane: Lessons from advanced imaging methods. Immunology 131:1–8.
- Pabst G, Danner S, Podgornik R, Katsaras J. 2007. Entropy-driven softening of fluid lipid bilayers by alamethicin. Langmuir 23:11705–11711.
- Palmer M, Valeva A, Kehoe M, Bhakdi S. 1995. Kinetics of streptolysin-O self-assembly. Eur J Biochem 231:388–395.
- Pantazaka E, Taylor CW. 2011. Differential distribution, clustering, and lateral diffusion of subtypes of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 286:23378–23387.
- Papo N, Shai Y. 2003. Exploring peptide membrane interaction using surface plasmon resonance: Differentiation between pore formation versus membrane disruption by lytic peptides. Biochemistry 42:458–466.
- Pastor RW, Venable RM, Feller SE. 2002. Lipid bilayers, NMR relaxation, and computer simulations. Acc Chem Res 35:438–446.
- Periasamy N, Bicknese S, Verkman AS. 1996. Reversible photobleaching of fluorescein conjugates in air-saturated viscous solutions: Singlet and triplet state quenching by tryptophan. Photochem Photobiol 63:265–271.
- Peters R, Cherry RJ. 1982. Lateral and rotational diffusion of bacteriorhodopsin in lipid bilayers: Experimental test of the Saffman-Delbrück equations. Proc Natl Acad Sci USA 79:4317–4321.
- Pinaud F, Clarke S, Sittner A, Dahan M. 2010. Probing cellular events, one quantum dot at a time. Nat Methods 7:275–285.
- Pokorny A, Almeida PFF. 2004. Kinetics of dye efflux and lipid flip-flop induced by δ-lysin in phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, α-helical peptides. Biochemistry 43:8846–8857.
- Pomorski T, Hrafnsdottir S, Devaux PF, van Meer G. 2001. Lipid distribution and transport across cellular membranes. Semin Cell Dev Biol 12:139–148.
- Pridmore CJ, Mosely JA, Rodger A, Sanderson JM. 2011. Acyl transfer from phosphocholine lipids to melittin. Chem Commun 47:1422–1424.
- Przybylo M, Sýkora J, Humpolíčková J, Benda A, Zan A, Hof M. 2006. Lipid diffusion in giant unilamellar vesicles is more than 2 times faster than in supported phospholipid bilayers under identical conditions. Langmuir 22:9096–9099.
- Pu M, Fang X, Redfield AG, Gershenson A, Roberts MF. 2009. Correlation of vesicle binding and phospholipid dynamics with phospholipase C activity: Insights into phosphatidylcholine activation and surface dilution inhibition. J Biol Chem 284:16099–16107.
- Qian S, Heller WT. 2011. Peptide-induced asymmetric distribution of charged lipids in a vesicle bilayer revealed by small-angle neutron scattering. J Phys Chem B 115:9831–9837.
- Qian S, Wang W, Yang L, Huang HW. 2008. Structure of the alamethicin pore reconstructed by x-ray diffraction analysis. Biophys J 94:3512–3522.
- Ramadurai S, Holt A, Krasnikov V, van den Bogaart G, Killian JA, Poolman B. 2009. Lateral diffusion of membrane proteins. J Am Chem Soc 131:12650–12656.
- Rapson AC, Hossain MA, Wade JD, Nice EC, Smith TA, Clayton AHA, 2011. Structural dynamics of a lytic peptide interacting with a supported lipid bilayer. Biophys J 100:1353–1361.
- Reddy K, Yedery R, Aranha C. 2004. Antimicrobial peptides: Premises and promises. Int J Antimicrob Ag 24:536–547.
- Risselada HJ, Marrink SJ. 2009. Curvature effects on lipid packing and dynamics in liposomes revealed by coarse grained molecular dynamics simulations. Phys Chem Chem Phys 11:2056–2067.
- Rodger A, Rajendra J, Marrington R, Ardhammar M, Nordén B, Hirst JD, 2002. Flow oriented linear dichroism to probe protein orientation in membrane environments. Phys Chem Chem Phys 4:4051–4057.
- Rolfe DJ, McLachlan CI, Hirsch M, Needham SR, Tynan CJ, Webb SED, 2011. Automated multidimensional single molecule fluorescence microscopy feature detection and tracking. Eur Biophys J 40:1167–1186.
- Ross M, Gerke V, Steinem C. 2003. Membrane composition affects the reversibility of annexin A2t binding to solid supported membranes: A QCM study. Biochemistry 42:3131–3141.
- Rothman JE, Dawidowicz EA. 1975. Asymmetric exchange of vesicle phospholipids catalyzed by the phosphatidylcholine exchange protein: Measurement of inside-outside transitions. Biochemistry 14:2809–2816.
- Rothman JE, Tsai DK, Dawidowicz EA, Lenard J. 1976. Transbilayer phospholipid asymmetry and its maintenance in the membrane of influenza virus. Biochemistry 15:2361–2370.
- Roullier V, Clarke S, You C, Pinaud F, Gouzer GG, Schaible D, 2009. High-affinity labeling and tracking of individual histidine-tagged proteins in live cells using Ni2+ tris-nitrilotriacetic acid quantum dot conjugates. Nano Lett 9:1228–1234.
- Ryba NJ, Marsh D. 1992. Protein rotational diffusion and lipid/protein interactions in recombinants of bovine rhodopsin with saturated diacylphosphatidylcholines of different chain lengths studied by conventional and saturation-transfer electron spin resonance. Biochemistry 31:7511–7518.
- Saffman PG, Delbrück M. 1975. Brownian motion in biological membranes. Proc Natl Acad Sci USA 72:3111–3113.
- Salnikov ES, Bechinger B. 2011. Lipid-controlled peptide topology and interactions in bilayers: Structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys J 100:1473–1480.
- Salnikov E, Aisenbrey C, Vidovic V, Bechinger B. 2010. Solid-state NMR approaches to measure topological equilibria and dynamics of membrane polypeptides. Biochim Biophys Acta 1798:258–265.
- Sanyal S, Menon AK. 2009. Flipping lipids: Why an' what's the reason for? ACS Chem Biol 4:895–909.
- Sapay N, Bennett WFD, Tieleman DP. 2010. Molecular simulations of lipid flip-flop in the presence of model transmembrane helices. Biochemistry 49:7665–7673.
- Schneider SW, Larmer J, Henderson RM, Oberleithner H. 1998. Molecular weights of individual proteins correlate with molecular volumes measured by atomic force microscopy. Pflüg Arch Eur J Phy 435:362–367.
- Scheidt HA, Huster D, Gawrisch K. 2005. Diffusion of cholesterol and its precursors in lipid membranes studied by 1H pulsed field gradient magic angle spinning NMR. Biophys J 89:2504–2512.
- Schwarz G, Robert CH. 1990. Pore formation kinetics in membranes, determined from the release of marker molecules out of liposomes or cells. Biophys J 58:577–583.
- Schwarz G, Robert CH. 1992. Kinetics of pore-mediated release of marker molecules from liposomes or cells. Biophys Chem 42:291–296.
- Schwarz G, Gerke H, Rizzo V, Stankowski S. 1987. Incorporation kinetics in a membrane, studied with the pore-forming peptide alamethicin. Biophys J 52:685–692.
- Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462:55–70.
- Shaw JE, Epand RF, Epand RM, Li ZG, Bittman R, Yip CM. 2006. Correlated fluorescence-atomic force microscopy of membrane domains: Structure of fluorescence probes determines lipid localization. Biophys J 90:2170–2178.
- Singer SJ, Nicolson GL. 1972. The fluid mosaic model of the structure of cell membranes. Science 175:720–731.
- Skaug MJ, Faller R, Longo ML. 2011. Correlating anomalous diffusion with lipid bilayer membrane structure using single molecule tracking and atomic force microscopy. J Chem Phys 134:215101.
- Sonnleitner A, Schütz GJ, Schmidt TH. 1999. Free Brownian motion of individual lipid molecules in biomembranes. Biophys J 77:2638–2642.
- Su M, He CF, West CA, Mentzer SJ. 2001. Cytolytic peptides induce biphasic permeability changes in mammalian cell membranes. J Immunol Methods 252:63–71.
- Svensson FR, Lincoln P, Nordén B, Esbjörner EK. 2011. Tryptophan orientations in membrane-bound gramicidin and melittin – a comparative linear dichroism study on transmembrane and surface-bound peptides. Biochim Biophys Acta 1808:219–228.
- Swaminathan R, Hoang CP, Verkman AS. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J 72:1900–1907.
- Thomas CJ, Anbazhagan V, Ramakrishnan M, Sultan N, Surolia I, Swamy MJ. 2003. Mechanism of membrane binding by the bovine seminal plasma protein, PDC-109: A surface plasmon resonance study. Biophys J 84:3037–3044.
- Tieleman DP, Marrink S-J. 2006. Lipids out of equilibrium: Energetics of desorption and pore mediated flip-flop. J Am Chem Soc 128:12462–12467.
- Uchida K, Emoto K, Daleke DL, Inoue K, Umeda M. 1998. Induction of apoptosis by phosphatidylserine. J Biochem 123:1073–1078.
- Ulrich K, Sanders M, Grinberg F, Galvosas P, Vasenkov S. 2008. Application of pulsed field gradient NMR with high gradient strength for studies of self-diffusion in lipid membranes on the nanoscale. Langmuir 24:7365–7370.
- Vad BS, Bertelsen K, Johansen CH, Pedersen JM, Skrydstrup T, Nielsen NC, 2010. Pardaxin permeabilizes vesicles more efficiently by pore formation than by disruption. Biophys J 98:576–585.
- Valentine CD, Haggie PM. 2011. Confinement of β1- and β2-adrenergic receptors in the plasma membrane of cardiomyocyte-like H9c2 cells is mediated by selective interactions with PDZ domain and A-kinase anchoring proteins but not caveolae. Mol Biol Cell 22:2970–2982.
- van den Bogaart G, Hermans N, Krasnikov V, Poolman B. 2007. Protein mobility and diffusive barriers in Escherichia coli: Consequences of osmotic stress. Mol Microbiol 64:858–871.
- Veatch SL, Keller SL. 2005. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys Rev Lett 94:148101.
- Verhulst PM, Holthuis JCM, Pomorski TG. 2012. Flip or flop: Mechanism and (patho)physiology of P4-ATPase-catalyzed lipid transport. In: Devaux PF, Herrmann A, editors. Transmembrane dynamics of lipids. Hoboken, NJ: Wiley. pp 149–170.
- Volinsky R, Cwiklik L, Jurkiewicz P, Hof M, Jungwirth P, Kinnunen PKJ. 2011. Oxidized phosphatidylcholines facilitate phospholipid flip-flop in liposomes. Biophys J 101:1376–1384.
- Wacklin HP. 2011. Composition and asymmetry in supported membranes formed by vesicle fusion. Langmuir 27:7698–7707.
- Wacklin HP, Tiberg F, Fragneto G, Thomas RK. 2007. Distribution of reaction products in phospholipase A2 hydrolysis. Biochim Biophys Acta 1768:1036–1049.
- Wagner ML, Tamm LK. 2000. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: Silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys J 79:1400–1414.
- Wang T, Li D, Lu X, Khmaladze A, Han X, Ye S, 2011. Single lipid bilayers constructed on polymer cushion studied by sum frequency generation vibrational spectroscopy. J Phys Chem C 115:7613–7620.
- Wang TY, Silvius JR. 2000. Different sphingolipids show differential partitioning into sphingolipid/cholesterol-rich domains in lipid bilayers. Biophys J 79:1478–1489.
- Weiss M. 2004. Challenges and artifacts in quantitative photobleaching experiments. Traffic 5:662–671.
- Wimley WC. 2010. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol 5:905–917.
- Won S, Kim H-D, Kim J-Y, Lee B-C, Chang S, Park C-S. 2010. Movements of individual BKCa channels in live cell membrane monitored by site-specific labeling using quantum dots. Biophys J 99:2853–2862.
- Wu Y, Huang HW, Olah GA. 1990. Method of oriented circular dichroism. Biophys J 57:797–806.
- Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. 2001. Barrel-stave model or toroidal model?: A case study on melittin pores. Biophys J 81:1475–1485.
- Yang P, Ramamoorthy A, Chen Z. 2011. Membrane orientation of MSI-78 measured by sum frequency generation vibrational spectroscopy. Langmuir 27:7760–7767.
- Yengo C, Berger CL. 2010. Fluorescence anisotropy and resonance energy transfer: Powerful tools for measuring real time protein dynamics in a physiological environment. Curr Opin Pharmacol 10:731–737.
- Zhang L, Granick S. 2005. Lipid diffusion compared in outer and inner leaflets of planar supported bilayers. J Chem Phys 123:211104.
- Zhang L, Booth CA, Stroeve P. 2000. Phosphatidylserine/cholesterol bilayers supported on a polycation/alkylthiol layer pair. J Colloid Interf Sci 228:82–89.
- Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. 1991. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol 173:2026–2034.