Abstract
Despite detailed genetic and mutagenic analysis and a recent high-resolution structure of a bacterial member of the nucleobase-ascorbate transporter (NAT) family, understanding of the mechanism of action of eukaryotic NATs is limited. Preliminary studies successfully expressed and purified wild-type UapA to high homogeneity; however, the protein was extremely unstable, degrading almost completely after 48 h at 4°C. In an attempt to increase UapA stability we generated a number of single point mutants (E356D, E356Q, N409A, N409D, Q408E and G411V) previously shown to have reduced or no transport activity, but correct targeting to the membrane. The mutant UapA constructs expressed well as GFP fusions in Saccharomyces cerevisiae and exhibited similar fluorescent size exclusion chromatography (FSEC) profiles to the wild-type protein, following solubilization in 1% DDM, LDAO or OM + 1 mM xanthine. In order to assess the relative stabilities of the mutants, solubilized fractions prepared in 1% DDM + 1 mM xanthine were heated at 45°C for 10 min prior to FSEC. The Q408E and G411V mutants gave markedly better profiles than either wild-type or the other mutants. Further FSEC analysis following solubilization of the mutants in 1% NG ± xanthine confirmed that G411V is more stable than the other mutants, but showed that Q408E is unstable under these conditions. G411V and an N-terminally truncated construct G411VΔ1-11 were submitted to large-scale expression and purification. Long-term stability analysis revealed that G411VΔ1-11 was the most stable construct and the most suited to downstream structural studies.
Introduction
The Nucleobase-Ascorbate Transporter (NAT) family, also known as the nucleobase-cation symporter-2 (NCS2) family, comprises a widespread group of transmembrane transporters with roles in the uptake of important cellular metabolites (de Koning and Diallinas Citation2000, Diallinas and Gournas Citation2008, Gournas et al. Citation2008). Across the family there are some key differences in substrate profile and mechanism of action. The mammalian NATs function to transport L-ascorbic acid or nucleobases (xanthine, hypoxanthine, guanine, thymine or uracil) by a Na+ dependent symport mechanism (Tsukaguchi et al. Citation1999, Yamamoto et al. Citation2010). In contrast the non-mammalian NATs transport the nucleobases xanthine, uric acid or uracil, via an H+ dependent symport mechanism (Diallinas and Gournas Citation2008, Gournas et al. Citation2008).
In common with many other transporter families the NATs are predicted to contain 12 or 14 transmembrane domains (TMDs) together with intracellular N and C-termini (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008a, Lu et al. Citation2011). All NAT members contain two specific and functionally important motifs, the NAT signature motif, [A/G/S]-[Q/E/P]-N-X-G-X-X-X-X-T-[R/K/G] (where X is a hydrophobic amino acid residue) (Diallinas and Gournas Citation2008, Gournas et al. Citation2008) and the QH motif (Pantazopoulou and Diallinas Citation2006) located in TMD1.The NAT signature motif was initially thought to be located in a loop region just upstream of TMD9 on the basis of extensive biochemical and genetic analysis (Diallinas and Gournas Citation2008, Gournas et al. Citation2008) of UapA, a uric acid-xanthine transporter from the filamentous fungus Aspergillus nidulans (Diallinas and Scazzocchio Citation1989). This finding was supported by research on XanQ, the xanthine transporter from Escherichia coli (Karatza and Frillingos Citation2005, Georgopoulou et al. Citation2010). However, the recently published structure of the bacterial homologue UraA, a uracil transporter from E. coli in complex with uracil revealed that the NAT motif forms a half-helix which lies within TMD10 (Lu et al. Citation2011). Subsequent homology modeling revealed that the NAT motif of UapA and XanQ, and probably all NATs, forms part of TMD10 (Karena and Frillingos Citation2009, Amillis et al. Citation2011).
Residues within the NAT motif are known to have important roles in both transport function and substrate selectivity. Even the most conserved substitutions of some of the residues in this region have profound effects on transport activity although importantly they do not affect trafficking of the transporter to the membrane (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008b). Mutational analysis of the NAT motif of UapA highlighted a key role of Gln408 in substrate binding and transport kinetics, whereas the invariant residue Asn409 is essential for transport but does not have a role in substrate binding. In addition, residue Gly411 has been shown to be essential for transport kinetics of the protein, but may also have roles in substrate specificity (Koukaki et al. Citation2005). Similar results have been obtained for the equivalent residues of the E. coli xanthine transporter XanQ (Karatza et al. Citation2006, Papakostas et al. Citation2008, Georgopoulou et al. Citation2010, Mermelekas et al. Citation2010).
The recent structure of the uracil/H+symporter UraA from E. coli provided unprecedented detail into the mechanism of NAT proteins and is in good agreement with many of the findings of genetic analysis of UapA and XanQ (Lu et al. Citation2011). In the UraA structure the Gly289 and Glu290 (positions 1 and 2 of the NAT motif respectively and equivalent to Ala407 and Gln408 of UapA) form direct interactions with the substrate (Lu et al. Citation2011) together with the completely conserved Glu241 and Phe73 (Glu356 and Phe155 in UapA). The H-bond network formed between uracil and the two Glu residues is particularly important as mutation of either residue results in a complete loss of uracil binding. Mutagenesis of the residues equivalent to Glu290 and Glu241 of UraA in UapA (Glu356 and Gln408, respectively) results in loss of transport activity or dramatic modification of substrate binding affinity (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008b), without affecting correct membrane localization, confirming the important functional role of these residues. Intriguingly, in contrast to data obtained in UapA or XanQ, the invariant Asn291 (found in position 3 of the NAT motif and equivalent to Asn409 in UapA) to Ala had little effect on uracil uptake activity (Lu et al. Citation2011). Indeed in the protein structure, this residue is found some distance from the uracil binding site. Recent evidence from the molecular modeling of UapA and XanQ suggested that the invariant Asn of the NAT signature motif interacts with the His residue in TMD1 (QH motif), and thus might indirectly affect the function, of at least some members of the NAT family (Amillis et al. Citation2011, Karena and Frillingos Citation2011).
One other key residue involved in substrate binding by UraA is His245 (Lu et al. Citation2011). Mutation of His245 to Ala results in a complete loss of uracil binding activity in UraA. Interestingly, both the uracil molecule in the substrate binding site of UraA and the His245 also form H-bond interactions with the sugar head group of a molecule of n-nonyl-β-D-glucopyranoside (NG), the detergent used for purification and crystallization (Lu et al. Citation2011). It is suggested that the detergent molecule stabilizes the uracil in the binding pocket and this in turn contributes to the stability of the whole protein. In UapA and a range of other NAT transporters, including XanQ and human SVCT1 and 2, His245 of UraA is replaced with Asp. This residue appears to be important for function in XanQ as replacement of the carboxyl group leads to loss of transporter function (Mermelekas et al. Citation2010). However, the exact role of this residue in NATs is unknown.
The high resolution structure of UraA highlighted both general features of NAT function as well as key molecular differences between the members of the NAT family, which may reflect differences in substrate specificity and mechanism of action. Molecular modeling of UapA and other NAT family members is useful for the rational identification of residues with key roles in transporter function. However, it is essential that we obtain structural information for other NAT proteins, and in particular eukaryotic members of the NAT family.
Previously we reported that it was possible to over express UapA in Saccharomyces cerevisiae as a fusion protein with a C-terminal GFP (Leung et al. Citation2010). While NATs are found in virtually every organism, one notable exception is S. cerevisiae. It was possible to exploit the lack of a NAT in this organism to show that the UapA-GFP was functionally expressed (Leung et al. Citation2010). Importantly, it was possible to isolate mg quantities of UapA which showed CD spectra characteristic of a highly α-helical protein. However, preliminary analysis indicated that the purified wild-type UapA was highly unstable, subject to dramatic degradation after only 48 h post isolation. Several UapA mutants of highly conserved residues have been previously identified with very low levels of transport activity but wild-type localization to the membrane (Diallinas and Gournas Citation2008). Here we have screened these mutants for improved stability compared to the wild-type UapA. We describe how by a combination of mutagenesis, N-terminal truncation and optimization of purification buffers, we have significantly improved the stability of UapA. The most stable construct generated by this study is highly suitable for structural studies.
Methods
Site-directed mutagenesis and generation of the N-terminal truncated construct
Mutants (E356D, E356Q, Q408E, N409A, N409D and G411V) of UapA () were generated using the QuikChange Mutagenesis kit (Stratagene) and the oligonucleotide primer pairs given in . The presence of the mutations was confirmed by DNA sequencing. An N-terminally truncated version of the G411V mutant lacking the N-terminal 11 residues was generated using the primers detailed in to give the construct G411VΔ1-11.
Table I. Oligonucleotide primers used in this study.
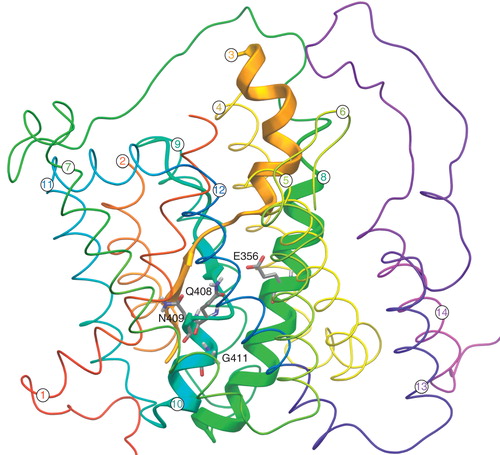
Figure 1. 3D modelled structure of UapA consisting of 14 α-helical TMDs and cytoplasmically located N- and C-termini (based on Amillis et al. Citation2011). TMDs 3, 8 and 10 predicated to be involved in substrate binding are indicated by the thick ribbons while all other residues are indicated by the thin ribbons. The residues relevant to this work (E356, Q408, N409, G411) are located on TMDs 8 and 10 and are shown as stick models. This figure is reproduced in color in the online version of Molecular Membrane Biology.
Expression of wild-type, mutant and truncated UapA constructs
All the UapA constructs described in the study were produced as C-terminal GFP fusion proteins using the pDDGFP-2 S. cerevisiae expression vector (Drew et al. Citation2008) as previously described (Leung et al. Citation2010). All the proteins were expressed in the pep4Δ deletion strain, FGY217 (Woolford et al. Citation1986). Preliminary analysis of expression of all the constructs was carried out in small scale cultures. Colonies for each individual construct were cultured in 10 ml URA media medium supplemented with 0.1% glucose and protein expression induced with a final concentration of 2% galactose for 22 h. The cell pellets were then harvested and the GFP emission was measured at 512 nm, following excitation at 488 nm in a microplate spectrofluorometer (SpectraMax) (Leung et al. Citation2010). The protein expression level was calculated from the relative fluorescence units (RFU), as described previously (Drew et al. Citation2008). Medium (1 l) and large (10 l) scale expression and membrane preparation were carried out as previously described (Leung et al. Citation2010).
Solubilization
Membranes containing 3 mg of total membrane protein were solubilized in solubilization buffer (1 × PBS, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM xanthine and 2 complete protease inhibitor tablets (Roche) per 250 ml supplemented with detergent [either 1% (w/v) n-Dodecyl-β-D-Maltoside (DDM), 1% n-Dodecyl-N,N-Dimethylamine-N-Oxide (LDAO), 1% n-Nonyl-β-D-Glucoside (NG) or 2% n-Octyl-β-D-Maltopyranoside (OM)] with constant stirring at 4°C for 1 h. Unsolubilized membrane was pelleted by centrifugation at 150,000 g and 4°C for 45 min.
FSEC and heated FSEC
Fluorescent size exclusion chromatography (FSEC) of the UapA-GFP fusion proteins was carried out on the mutant and wild-type UapA-GFPs as described by Kawate and Gouaux (Citation2006) following solubilization in 1% DDM, 1% LDAO or 2% OM. Heated FSEC was carried out in the same way with the exception that the solubilized protein was heated prior to loading onto the SEC column (Superose 6, GE Biosciences). In order to identify the optimal temperature to use for pretreatment of the solubilized UapA-GFP, a range of temperatures was tested. The membranes from a medium scale culture were solubilized for 1 h in 1% DDM + 1 mM xanthine. Aliquots of the solubilized material were heated at 40, 45 or 50°C for 10 min, cooled on ice for 2 min then centrifuged at 18,000 g for 10 min at 4°C prior to loading onto the SEC column. The fractions were collected in a 96-well plate format fraction collector and the fluorescence of all the samples assessed. The data obtained was then plotted using GraphPad Prism 5. The optimal temperature where it was possible to observe a significant reduction in the monodispersity of the fusion protein peak but not a complete loss for UapA-GFP was 45°C. Therefore all the modified constructs were incubated at 45°C for 10 min and the FSEC profiles compared to that of wild-type UapA treated in the same way. The mutant and wild-type UapA-GFPs were also submitted to FSEC following solubilization in 1% NG ± 1 mM xanthine. The G411VΔ1-11 construct was also analyzed by heated FSEC following solubilization in 1% DDM and by FSEC following solubilization in 1% NG ± 1 mM xanthine.
Purification
The wild-type and modified constructs were all isolated as described previously (Leung et al. Citation2010) with minor modifications. In brief, the solubilized protein sample was mixed with Ni2+-NTA superflow equilibrated with Buffer A (1 × PBS, pH 7.5, 150 mM NaCl, 10% glycerol, 10 mM imidazole, 1 mM xanthine and 0.03% DDM) for 5 h at 4°C with slow stirring. The resin was washed with Buffer A with increasing concentrations of imidazole (up to 30 mM). Bound protein was eluted with Buffer B (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20% glycerol, 1 mM xanthine 250 mM imidazole, and 0.03% DDM). The GFP tag was removed from the protein with an equimolar amount of His-tagged TEV protease and the protein dialyzed overnight in Buffer B without imidazole followed by a separation on a 5 ml His-trap column (GE Biosciences). The untagged UapA was in the flowthrough while the His tagged GFP and His-tagged TEV were bound to the column. Following concentration of the isolated UapA to 0.5 ml using a 50 kDa molecular weight cut-off filter and centrifugation at 18,000 g to remove aggregates the protein sample was loaded onto a Superdex 200 10/300 column equilibrated with 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5% glycerol, 500 μM xanthine, and 0.03% DDM. The protein fractions were collected and analyzed by SDS-PAGE.
Long-term stability analysis
Long-term stability of pure UapA was assessed following incubation at 4°C or 20°C for a period of 6 days. Aliquots were collected on days 0, 1, 2, 3, and either 5 or 6. The fractions were stored in SDS running buffer at −20°C prior to SDS-PAGE analysis.
SDS-PAGE and in-gel fluorescence analysis
The protein samples were separated on Novex 12% Tris-Gly gels (Invitrogen). The gels were rinsed with dH2O and the protein bands visualized using Coomassie Blue stain following detection of the fluorescent bands with a LAS-1000-3000 charge-coupled device (CCD) imaging system (Fujifilm). The gel was exposed to blue light (460 nm) for 20 sec with a cut-off filter of 515 nm.
Results
Expression and preliminary stability analysis of mutant versions of UapA
We have chosen to test wild-type and several mutant versions of UapA (E356D, E356Q, Q408E, N409A, N409D and G411V, ). Our rationale for the selection of UapA mutants was based on previous in vivo observations in A. nidulans, which showed that the selected UapA mutants localize to the plasma membrane, as judged microscopically using GFP-tagged versions of the mutant transporters (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008b). All mutants selected also showed dramatically reduced ability to transport xanthine or uric acid. Of these, Q408E has lost its ability for substrate binding at 25°C (Koukaki et al. Citation2005), whereas the E356D binds substrate with high affinity but has reduced transport activity (Papageorgiou et al. Citation2008b, V. Kosti, E. Mikros and G. Diallinas, unpublished observations). The rest of the mutants seem to be incapable of transport activity due to conformational changes associated with substrate translocation, rather than substrate binding (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008b).
Measurement of the relative fluorescence units (RFU) of cultures expressing the different mutants indicated that they were produced to similar levels as the wild-type (1.9 mg/l) UapA-GFP (). In-gel fluorescence revealed the presence of a single fluorescent band, of approximately 75 kDa for both the wild-type UapA and each mutant fusion protein expressed (data not shown). FSEC of the mutants solubilized in 1% DDM + 1 mM xanthine, 1% LDAO + 1 mM xanthine or 2% OM + 1 mM xanthine gave very similar profiles to the wild-type UapA solubilized in the same conditions (see for representative examples). The DDM and OM solubilized GFP-fusion proteins gave single monodispersed peaks (eluting at about fractions 35–40) corresponding to the UapA-GFP fusions while the proteins solubilized in LDAO gave broad polydispersed peaks and substantial aggregation peaks (eluting at about fraction 8). The profiles indicated that the mutants were as stable as the wild-type in the three detergents under the conditions tested, and that all the proteins were more stable in DDM and OM than in LDAO. This analysis did not however allow the identification of UapA mutants that are more stable than wild-type.
Figure 2. FSEC analysis of wild-type UapA and the N409A mutant following solubilization in (a) and (b) 1% DDM, (c) and (d) 1% LDAO and (e) and (f) 2% OM, all in the presence of 1 mM xanthine. Of the six mutants generated the FSEC profiles for only N409A are shown. However, almost identical results were obtained for all the other mutants.
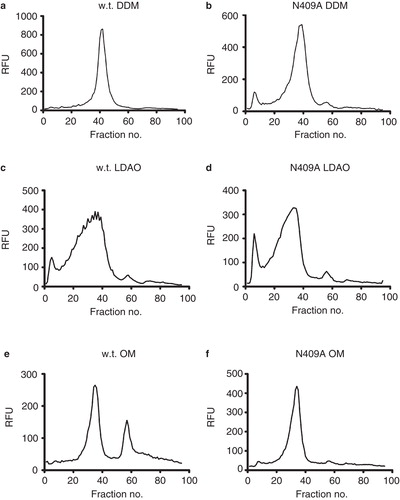
Table II. Summary of the relative stabilities of the different UapA constructs.
Heat FSEC provides a means of identifying more stable mutants
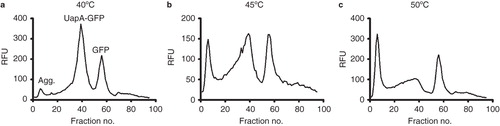
A method was required which stressed the protein samples to just the right amount to allow distinction between the relative stabilities of the mutant UapA-GFP constructs. We used a modified FSEC method where protein solubilized in 1% DDM + 1 mM xanthine was heated prior to loading on the SEC column. The ideal temperature and time of incubation (45°C and 10 min) was identified as causing some, but not a total, loss of monodispersity of the wild-type UapA ().
Figure 3. Heated FSEC trials of wild-type UapA to identify the optimum temperature for pretreatment of the solubilized protein prior to separation on a Superose 6 column. The solubilized protein samples were incubated at (a) 40°C, (b) 45°C or (c) 50°C for 10 min. The peaks corresponding to aggregated protein (void), the UapA-GFP fusion protein and free GFP are indicated in (a).
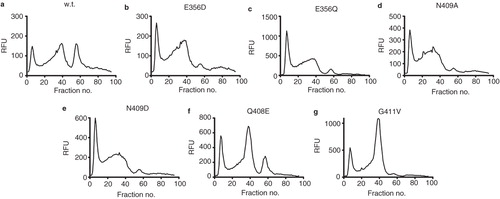
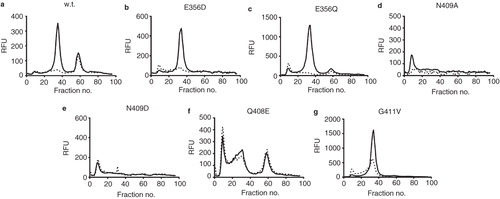
Heated FSEC of the mutants solubilized in 1% DDM + 1 mM xanthine following heating at 45°C for 10 min () revealed that the Q408E and G411V mutants had a significantly improved profile compared to wild-type with a much greater proportion of protein present in the monodispersed fusion protein peak (fraction 40) than the wild-type. Both the mutants express at slightly higher levels than the wild-type which may explain some but certainly not all of this increase ().
Figure 4. Heated FSEC analysis of (a) wild-type, (b) E356D, (c) E356Q, (d) N409A, (e) N409D, (f) Q408E and (g) G411V constructs of UapA following solubilization in 1% DDM + 1 mM xanthine.
Xanthine has a marked stabilization effect on UapA solubilized in 1% NG
Further analysis investigated the effects of solubilization in 1% NG ± 1 mM xanthine on the FSEC profiles of G411V and Q408E compared to wild-type. In this case the samples were not heated prior to loading on the SEC column. The wild type UapA in NG in the absence of xanthine shows virtually no recovery of fusion protein with only a small peak visible at about fraction number 60 corresponding to free GFP (). In stark contrast, solubilization in NG + 1 mM xanthine resulted in a single monodispersed peak indicating that xanthine has a major stabilizing effect on the wild-type UapA under these conditions. The Q408E mutants yielded poor FSEC profiles following solubilization in both NG only and NG + 1 mM xanthine (). This result is in agreement with earlier studies demonstrating that this mutant exhibits very low substrate binding in vivo (Papageorgiou et al. Citation2008b). In contrast, the G411V mutant solubilized in 1% NG + 1 mM xanthine yielded a single monodispersed peak similar to that seen for the wild-type UapA under the same conditions although the peak intensity was much higher. Taken together, the FSEC analyses under a range of conditions strongly indicated that the G411V was the most stable of the mutants tested ().
Figure 5. FSEC analysis of (a) wild-type, (b) E356D, (c) E356Q, (d) N409A, (e) N409D, (f) Q408E and (g) G411V constructs of UapA following solubilization in 1% NG (dashed lines) and 1% NG + 1 mM xanthine (solid lines).
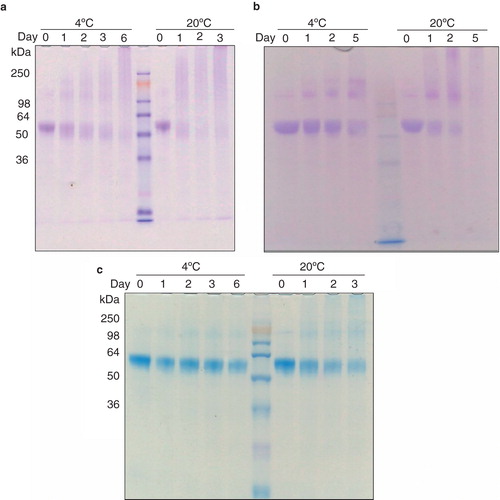
Removal of N-terminal residues further increases the stability of the G411V
In an effort to increase the stability of the protein further we identified the presence of a possible disordered region at the N-terminal end of the protein using the Regional Order Neural Network (RONN; Yang et al. Citation2005). A truncated construct of the G411V mutant was generated lacking the first 11 amino acids, G411VΔ1-11. Both FSEC of G411VΔ1-11 following solubilization in 1% NG + 1 mM xanthine and hFSEC of the construct following solubilization in 1% DDM + 1 mM xanthine resulted in almost identical profiles to the G411V mutant under the same conditions. These results indicate that the G411VΔ1-11 is at least as stable as G411V (). The wild-type UapA, G411V andG411VΔ1-11 constructs were all purified in DDM to high homogeneity as assessed by SDS-PAGE analysis (; Day 0) and as largely monodispersed samples as assessed by SEC (results not shown). The yields of the proteins differed markedly: wild-type, 0.15 mg/l; G411V 0.28 mg/l; G411VΔ1-11, 0.42 mg/l. Long-term stability analysis of the wild-type UapA assessed at both 4°C and 20°C revealed the highly unstable nature of the wild-type protein (). As mentioned at day 0, the purified wild-type UapA is observed as a single band of approximately 55 kDa. However, following 24 h incubation at 20°C the protein is visible only as a highly diffuse smear. The protein is slightly more stable at 4°C with some protein still visible as a single band after 24 h, but after 48 h almost all of the protein is visible as a smear (). The G411V is more stable, and is observable as a clear single band after 48 h incubation at 4°C. Some of the protein is also still present after incubation at 20°C for 24 h (). The G411VΔ1-11 shows the greatest stability with a single band visible six days after incubation at 4°C and although the amount of protein has significantly dropped there is still a single band visible after three days incubation at 20°C (; ).
Figure 6. Long-term stability analysis of purified (a) wild-type, (b) G411V and (c) G411VΔ1-11 following incubation at 4°C and 20°C for several days. The lanes containing the molecular weight markers are indicated. This Figure is reproduced in color in the online version of Molecular Membrane Biology.
Discussion
The stability of a membrane protein in solution is of fundamental importance to downstream functional and structural characterization. Previously we had shown that we could express the UapA in S. cerevisiae as a GFP fusion protein in a functional form (Leung et al. Citation2010). Although it proved possible to isolate wild-type UapA in amounts sufficient for further analysis the poor stability precluded submission to crystallization trials. In an attempt to increase the stability of the UapA, we generated a number of single point mutants which had previous been identified as showing reduced or no transporter activity but trafficked to the plasma membrane suggesting correct folding (Koukaki et al. Citation2005, Papageorgiou et al. Citation2008b).
While these mutants all expressed well in S. cerevisae as GFP fusions we were faced with the problem of how to effectively screen their relative stability compared to wild-type UapA without having to carry out large scale expression and purification. The optimal means of assessing stability of mutants compared to wild-type is to carry out functional analysis of the proteins at elevated temperatures. This works particularly well for G-protein coupled receptors (Serrano-Vega et al. Citation2008, Singh et al. Citation2010) as their function can be readily assessed in crude solubilized fractions allowing identification of comparatively more stable mutants at an early stage in the process. With transporters this is more challenging since assessment of the function of transporters requires reconstitution into liposomes, something only possible with purified sample. In addition, the mutant UapA constructs generated as part of this study have previously been shown to have low or no transporter activity.
We explored the use of FSEC (Kawate and Gouaux Citation2006) as a means to analyze our mutants. This technique allows characterization of crude solubilized protein samples. Initial attempts using standard FSEC together with protein detergents with different properties including zwitterionic and non-ionic head groups (DDM, LDAO) and different lengths of alkyl chains (DDM, OM) were unable to distinguish the relative stabilities of the wild-type and mutant UapA constructs. Therefore we modified the method slightly to incorporate an incubation step at elevated temperature prior to loading the samples onto the SEC column. It was necessary to experimentally determine the optimum incubation temperature, that is, a temperature which resulted in some, but not a complete, loss of monodispersed protein. In the case of wild-type UapA solubilized in 1% DDM + 1 mM xanthine this was 45°C. This heated FSEC method is likely to be suitable for analysis of a range of other solubilized membrane proteins however it may be necessary to perform preliminary screening to identify the optimal temperature at which to pretreat the samples.
The recent structure of the bacterial NAT protein, UraA, was crystallized as a complex with the bound substrate, uracil, in the short-chain detergent NG (Lu et al. Citation2011). The short chain length of NG together with the monosaccharide head group results in a comparatively small micelle (Robinson et al. Citation2008) which although advantageous for crystallization often causes dramatic destabilization of membrane proteins. In the case of the UraA in the absence of uracil, exchange into NG caused almost complete aggregation of the protein (Lu et al. Citation2011). In contrast, the combination of uracil and NG resulted in a protein stable enough to yield well diffracting crystals. The precise molecular reason for this was revealed by the high resolution structure which showed that an NG molecule binds on top of the uracil within the binding pocket forming key H-bonds with both the substrate and the transporter. It is thought that these interactions have the dual effects of stabilizing the uracil in the binding pocket and forming stabilizing interactions with the two main domains of the UraA.
In order to investigate whether UapA could be stabilized in a similar way we submitted all our mutants and the wild-type to FSEC with no prior heating following solubilization in either NG only or NG + 1 mM xanthine. Solubilization in NG only resulted in either very low or no recovery of monodispersed protein for all the mutants and the wild-type UapA. The clear exception to this was G411V. The addition of xanthine had a dramatic effect on the recovery of monodispersed protein in the case of the wild-type, E356D, E356Q and G411V. It is not entirely clear why this effect of xanthine is observed for these constructs and not the others. It may be that the other mutants show a dramatic reduction or a loss of function not simply because they no longer transport, but because they exhibit no or very low xanthine binding, as is the case for Q408E in vivo (Papageorgiou et al. Citation2008b). Alternatively, for mutants N409A and N409D which retain high-affinity binding for substrates (Koukaki et al. Citation2005), it may be that the introduction of these mutations allows binding of xanthine but causes subtle conformational changes in the substrate binding pocket of the protein. Such changes in turn may prevent the formation of key stabilizing H-bonds between a molecule of NG and UapA as observed between H245 and NG in the UraA structure (Lu et al. Citation2011).
Overall the results clearly demonstrated that G411V was the most stable mutant and this protein was subsequently purified and assessed for long-term stability. This analysis showed that there was a dramatic improvement in stability of the single point mutant compared to the wild-type UapA. In the UraA structure, the uracil binds into a pocket just above the half helix of TMD10 on the cytoplasmic side of the membrane. G293 (equivalent to G411 in UapA) is located roughly half way along the half helix, and is likely to impart a certain amount of flexibility to this region of the protein important for transport function. This, taken together with the data presented here and earlier findings that the G411V mutant exhibits a total loss of transport activity but wild-type binding, strongly indicates that a reduction in flexibility in this region caused by the presence of valine is responsible for the increase in stability seen in this mutant. Stability of the UapA was further improved by the removal of the N-terminal 11 amino acid residues predicted to be a source of significant structural flexibility.
In conclusion, it has proved possible to significantly improve the stability of UapA based on earlier studies identifying constructs with reduced or no functional activity, but correct plasma membrane localization, and thus proper folding. The resultant protein is much more suitable for downstream structural studies. This study highlights the great potential of combining genetic and biochemical ‘refinements’, easily achievable in model fungal systems, particularly for studying challenging proteins such as eukaryotic transporters.
Acknowledgements
This research was funded by the European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement, no. HEALTH-F4-2007-201924, EDICT Consortium.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Amillis S, Kosti V, Pantazopoulou A, Mikros E, Diallinas G. 2011. Mutational analysis and modeling reveal functionally critical residues in transmembrane segments 1 and 3 of the UapA transporter. J Mol Biol 411:567–580.
- de Koning H, Diallinas G. 2000. Nucleobase transporters (review). Mol Membr Biol 17:75–94.
- Diallinas G, Gournas C. 2008. Structure-function relationships in the nucleobase-ascorbate transporter (NAT) family: Lessons from model microbial genetic systems. Channels (Austin) 2:363–372.
- Diallinas G, Scazzocchio C. 1989. A gene coding for the uric acid-xanthine permease of Aspergillus nidulans: Inactivational cloning, characterization, and sequence of a cis-acting mutation. Genetics 122:341–350.
- Drew D, Newstead S, Sonoda Y, Kim H, Heijne von G, Iwata S. 2008. GFP-based optimization scheme for the over expression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat Prot 3:784–798.
- Georgopoulou E, Mermelekas G, Karena E, Frillingos S. 2010. Purine substrate recognition by the nucleobase-ascorbate transporter signature motif in the YgfO xanthine permease: ASN-325 binds and ALA-323 senses substrate. J Biol Chem 285:19422–19433.
- Gournas C, Papageorgiou I, Diallinas G. 2008. The nucleobase-ascorbate transporter (NAT) family: Genomics, evolution, structure-function relationships and physiological role. Mol Biosyst 4:404–416.
- Karatza P, Frillingos S. 2005. Cloning and functional characterization of two bacterial members of the NAT/NCS2 family in Escherichia coli. Mol Membr Biol 22:251–261.
- Karatza P, Panos P, Georgopoulou E, Frillingos S. 2006. Cysteine-scanning analysis of the nucleobase-ascorbate transporter signature motif in YgfO permease of Escherichia coli: Gln-324 and Asn-325 are essential, and Ile-329-Val-339 form an alpha-helix. J Biol Chem 281:39881–39890.
- Karena E, Frillingos S. 2009. Role of intramembrane polar residues in the YgfO xanthine permease: HIS-31 and ASN-93 are crucial for affinity and specificity, and ASP-304 and GLU-272 are irreplaceable. J Biol Chem 284:24257–24268.
- Karena E, Frillingos S. 2011. The role of transmembrane segment TM3 in the xanthine permease XanQ of Escherichia coli. J Biol Chem 286:39595–39605.
- Kawate T, Gouaux E. 2006. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14:673–681.
- Koukaki M, Vlanti A, Goudela S, Pantazopoulou A, Gioule H, Tournaviti S, 2005. The nucleobase-ascorbate transporter (NAT) signature motif in UapA defines the function of the purine translocation pathway. J Mol Biol 350:499–513.
- Leung J, Karachaliou M, Alves C, Diallinas G, Byrne B. 2010. Expression and purification of a functional uric acid-xanthine transporter (UapA). Prot Express Purif 72:139–146.
- Lu F, Li S, Jiang Y, Jiang J, Fan H, Lu G, 2011. Structure and mechanism of the uracil transporter UraA. Nature 472:243–246.
- Mermelekas G, Georgopoulou E, Kallis A, Botou M, Vlantos V, Frillingos S. 2010. Cysteine-scanning analysis of helices TM8, TM9a, and TM9b and intervening loops in the YgfO xanthine permease: A carboxyl group is essential at ASP-276. J Biol Chem 285:35011–35020.
- Pantazopoulou A, Diallinas G. 2006. The first transmembrane segment (TMS1) of UapA contains determinants necessary for expression in the plasma membrane and purine transport. Mol Membr Biol 23:337–348.
- Papageorgiou I, de Koning HP, Soteriadou K, Diallinas G. 2008a. Kinetic and mutational analysis of the Trypanosoma brucei NBT1 nucleobase transporter expressed in Saccharomyces cerevisiae reveals structural similarities between ENT and MFS transporters. Int J Parasitol 38:641–653.
- Papageorgiou I, Gournas C, Vlanti A, Amillis S, Pantazopoulou A, Diallinas G. 2008b. Specific interdomain synergy in the UapA transporter determines its unique specificity for uric acid among NAT carriers. J Mol Biol 382:1121–1135.
- Papakostas K, Georgopoulou E, Frillingos S. 2008. Cysteine-scanning analysis of putative helix XII in the YgfO xanthine permease: ILE-432 and ASN-430 are important. J Biol Chem 283:13666–13678.
- Robinson AJ, Overy C, Kunji ERS. 2008. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc Natl Acad Sci USA 105:17766–17771.
- Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. 2008. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA 105:877–882.
- Singh S, Hedley D, Kara E, Gras A, Iwata S, Ruprecht J, 2010. A purified C-terminally truncated human adenosine A(2A) receptor construct is functionally stable and degradation resistant. Prot Express Purif 74:80–87.
- Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, 1999. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 399:70–75.
- Woolford CA, Daniels LB, Park FJ, Jones EW, van Arsdell JN, Innis MA. 1986. The PEP4 gene encodes an aspartyl protease implicated in the posttranslational regulation of Saccharomyces cerevisiae vacuolar hydrolases. Mol Cell Biol 6:2500–2510.
- Yamamoto S, Inoue K, Murata T, Kamigaso S, Yasujima T, Maeda J-Y, 2010. Identification and functional characterization of the first nucleobase transporter in mammals: Implication in the species difference in the intestinal absorption mechanism of nucleobases and their analogs between higher primates and other mammals. J Biol Chem 285:6522–6531.
- Yang ZR, Thomson R, McNeil P, Esnouf RM. 2005. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21:3369–3376.