Abstract
The increasing number of multidrug-resistant pathogenic microorganisms is a serious public health issue. Among the multitude of mechanisms that lead to multidrug resistance, the active extrusion of toxic compounds, mediated by MDR efflux pumps, plays an important role. In our study we analyzed the inhibitory capability of 26 synthesized zosuquidar derivatives on three ABC-type MDR efflux pumps, namely Saccharomyces cerevisiae Pdr5 as well as Lactococcus lactis LmrA and LmrCD. For Pdr5, five compounds could be identified that inhibited rhodamine 6G transport more efficiently than zosuquidar. One of these is a compound with a new catechol acetal structure that might represent a new lead compound. Furthermore, the determination of IC50 values for rhodamine 6G transport of Pdr5 with representative compounds reveals values between 0.3 and 0.9 μM. Thus the identified compounds are among the most potent inhibitors known for Pdr5. For the ABC-type efflux pumps LmrA and LmrCD from L. lactis, seven and three compounds, which inhibit the transport activity more than the lead compound zosuquidar, were found. Interestingly, transport inhibition for LmrCD was very specific, with a drastic reduction by one compound while its diastereomers showed hardly an effect. Thus, the present study reveals new potent inhibitors for the ABC-type MDR efflux pumps studied with the inhibitors of Pdr5 and LmrCD being of particular interest as these proteins are well known model systems for their homologs in pathogenic fungi and Gram-positive bacteria.
Introduction
The expression of multidrug resistance (MDR) efflux pumps that actively extrude a broad spectrum of structurally unrelated compounds can cause multidrug resistance of cancer cells or pathogenic microorganisms. This often circumvents an effective treatment of tumors as well as infections (Piddock Citation2006a, Citation2006b, Szakacs et al. Citation2006). The MDR efflux pumps comprise members of the ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the multidrug and toxic-compound extrusion (MATE) family, the small multidrug resistance (SMR) family and the resistance nodulation division (RND) family (for reviews see Gottesman et al. Citation2002, Piddock Citation2006b). In mammals (e.g., humans) members of the ABC-transporter superfamily are typically responsible for the development of a multidrug resistance phenotype (Gottesman et al. Citation2002). Multidrug resistant pathogenic yeast (such as Candida albicans), normally (over-) expresses ABC-type and/or MFS-type MDR efflux pumps (Sanglard et al. Citation2009, Morschhauser Citation2010). In contrast, bacteria usually rely on efflux pumps of the RND- and MFS-type, respectively (for a review see Piddock Citation2006b). However, in the last few years it has turned out that the role of ABC-type efflux pumps in bacterial MDR was underestimated. Especially in several pathogenic Gram-positive bacteria (e.g., Streptococcus pneumoniae), ABC-type efflux pumps seem to be responsible for the development of a MDR phenotype (Lubelski et al. Citation2007). Therefore, the development of inhibitors for ABC-type efflux pumps of pathogenic fungi and bacteria represents an important step to overcome MDR in clinically important pathogens. For the design of potential inhibitors zosuquidar, a third-generation P-glycoprotein inhibitor (Pfister et al. Citation1995) was chosen as lead structure. Based on this compound, 26 derivatives have been synthesized (see ) and tested on three well-characterized model systems for MDR-ABC-transporters. The studied efflux pumps were Pdr5 from S. cerevisiae as well as LmrA and LmrCD from L. lactis.
Figure 1. Summary of the chemical structures of the compounds used in this study. All compounds are derived from zosuquidar, a third-generation P-gp inhibitor.
The ABC-type efflux pump Pdr5 is a major component of the pleiotropic drug resistance (PDR) network of S. cerevisiae that provides resistance to a broad spectrum of structurally unrelated xenobiotics (Kolaczkowski et al. Citation1996, Egner et al. Citation1998, Golin et al. Citation2003). Pdr5 contains two nucleotide binding domains (NBDs) fueling active transport by catalyzing the hydrolysis of ATP as well as two transmembrane domains (TMDs) harboring a total of 12 predicted transmembrane α-helicies, which characterizes Pdr5 as a so-called full-size transporter. Furthermore, Pdr5, in addition to its significant sequence homology with ABC-type MDR efflux pumps of pathogenic fungi, can be functionally complemented by its C. albicans homolog Cdr1 (Prasad et al. Citation1995), the deletion of which renders its host hypersensitive to a variety of antifungal drugs (e.g., azoles, terbinafine and amorolfine for a review see Sanglard et al. Citation2009). The latter highlights that Pdr5 represents an excellent model system for the study of ABC-type MDR efflux pumps of pathogenic fungi.
Similar to Pdr5, also the ABC-type efflux pumps LmrA and LmrCD from L. lactis have been shown to transport a broad range of classic MDR efflux pump substrates (e.g., Hoechst 33342, daunomycin and BCECF-AM) at the expense of ATP-hydrolysis (van Veen et al. Citation1996, Citation1998, Citation2000, Lubelski et al. Citation2004, Citation2006a), although some controversy exists in the literature concerning the Hoechst 33342 transport of LmrA (van den Berg van Saparoea et al. Citation2005, Venter et al. Citation2008). The two lactococcal efflux pumps are so-called half size transporters, which contain an N-terminal TMD and a C-terminal NBD (van Veen et al. Citation1996, Lubelski et al. Citation2004). Therefore the functional unit of these transporters is a homodimer in the case of LmrA and a heterodimer for LmrCD (van Veen et al. Citation2000, Lubelski et al. Citation2004). Furthermore, it has been reported for LmrA that its expression in a human cell line confers an MDR phenotype with pharmacological characteristics similar to cells expressing P-glycoprotein. This, together with other experiments, led to the conclusion that LmrA and the P-glycoprotein are functionally interchangeable (van Veen et al. Citation1998). However, in the natural host L. lactis, LmrCD seems to be more important for the development of MDR, as a clear phenotype is observed for the deletion as well as the overexpression of LmrCD (Lubelski et al. Citation2006a). Based on these experiments, LmrCD seems to be the major MDR-transporter in L. lactis (Lubelski et al. Citation2006a). Apart from this finding, LmrCD homologs (50–60% amino acid identity) have been identified in Gram-positive pathogens that were reported to fulfill major roles in MDR in these in clinically relevant microorganisms (Lubelski et al. Citation2007). This emphasizes the suitability of LmrCD as a model system.
Materials and methods
Materials
Medium components were obtained from Carl Roth; oligomycin, rhodamine 6G. Hoechst 33342 and ethidium bromide were obtained from Sigma-Aldrich. The synthesized compounds and the dyes were dissolved in water, and oligomycin in dimethyl sulfoxide. For determination of the protein concentration, a Coomassie Plus Assay (Pierce) was used.
Strains
Saccharomyces cerevisiae: YALF-A1 (Ernst et al. Citation2008);
Lactococcus lactis: NZ 9000 ΔlmrA ΔlmrCD (van den Berg van Saparoea et al. Citation2005).
Synthesis of potential inhibitors
Guided by the lead structure of the multidrug-resistance reversal agent zosuquidar (1) (Pfister et al. Citation1995, Starling et al. Citation1997, Dantzig et al. Citation2001, Sorbera et al. Citation2003), a series of compounds was synthesized that were considered as potential inhibitors of the MDR ABC-transporters studied. Our strategy was based on modification or replacement of the dibenzosuberyl and the piperazine moieties, whereas the 2-propanol and the 5-hydroxyisoquinoline units were maintained, including the absolute configuration of the stereogenic carbinol center.
Aside from the known compounds 1, 2, and 4- 8, that were prepared according to known procedures (Fukazawa et al. Citation1990, Citation1992, Sircar et al. Citation1992, Pfister and Lynne Citation1994, Suzuki et al. Citation1997), a total amount of 20 new quinoline derivatives were synthesized (see ). Among them, compounds 3 and 9- 16 can be considered as closely related to 2 and 4- 8. This group of compounds comprises 4-aminopiperidines 11- 13, piperidines 9- 10 missing the second nitrogen atom, and piperazines 14- 16, wherein the diphenylmethyl motif was replaced by a mono-aromatic substituent. A second approach towards novel inhibitors was based on the replacement of the dibenzosuberyl moiety in 1 by acetal or thioacetal groups and resulted in the novel spirocyclic compounds 17- 27. The acetals 20 and 21 formed mixtures of diastereomers. All the compounds were obtained in enantiomerically pure form, the stereogenic carbinol center adopting (R) configuration; compounds 23 and 24 are a pair of enantiomerically pure diastereomers (Dittrich et al. Citation2012).
In vivo ethidium transport assay
Culture of L. lactis cells that expressed LmrA or the E512Q mutant of LmrA were cooled on ice for at least 15 min. Subsequently, cells from 1 ml culture volume were washed three times with de-energizing buffer (50 mM Hepes, pH 7.4, 5 mM deoxyglucose, and 40 μM ethidium bromide), and were kept on ice for 20 min. Cells were than washed five times with 50 mM Hepes pH 7.0 to remove the deoxyglucose. Cells were resuspended in 2 ml 50 mM Hepes pH 7.0 and divided into two parts. To one part, the assayed compound was added with a final concentration of 30 μM, and the other part was used for a control measurement (no compound added). The concentration of 30 μM for the assayed compound was chosen to pick up potential inhibitors with a weaker potency, because at this concentration the reference compound zosuquidar reduced the ethidium transport to approximately 50% of the value without the compound. The fluorescence of the ethidium-DNA-complex was monitored at excitation and emission wavelengths of 500 and 580 nm, respectively. The measurements were performed with a fluorometer (Fluorolog II Horiba) with a magnetic stirred holder at 30°C. After starting the measurement, cells were re-energized with 20 mM glucose. After 30 min, transport was finished and a stable fluorescence signal was reached. The final fluorescence intensity was taken for quantification of the transport activity. Control measurements were performed by the addition of water instead of glucose. The compounds did not change the final fluorescence intensity of cells expressing E512Q-LmrA.
Fluorescence microscopy
Lactococcus lactis cells, treated the same way as described (see previous section), were fixed for 20 min at −20°C with ethanol on cover slides coated with poly-L-lysine. After removal of ethanol and two washing steps with water, the cells were embedded in mowiol 4-88 and stored light protected. Cells were examined with a 60 × 1.3 numerical aperture Zeiss oil-immersion objective using a Zeiss LSM 510 Meta confocal microscope system. Ethidium was excited with a 488-nm argon laser with emission detection through the meta channel at 565–634 nm. All parameters (frame size, pixel time, HV value) were kept constant for all measurements.
Isolation of membrane vesicles from Lactococcus lactis
Lactococcus lactis cells harboring the plasmid pNHLmrA (for expression of LmrA) or pNSGA (pNZ8048 with LmrCD gene with a downstream region encoding Streptag II (Lubelski et al. Citation2004)) were cultivated at 30°C in M17 medium supplemented with 0.5% (w/v) glucose and 5 μg/ml chloramphenicol. Cells were grown to an OD660 of about 0.8 and expression of LmrA or LmrCD was induced by the addition of 25 μg/l Nisin (Fluka). After 2 h of expression, cells were harvested by centrifugation and stored at −80°C. The cell suspension was thawed and incubated with lysozyme (10 mg/ml) for 30 min at 30°C. Subsequently, the suspension was cooled on ice for 15 min and EDTA was added to a final concentration of 4 mM. Cells were lysed by passing them four times through a cooled TS Series Cell Disruptor (Constant Systems) at 2.5 kbar. Cell debris and intact cells were removed by two centrifugation steps (45 min at 13,000 g and 30 min at 20,000 g at 4°C). Membrane vesicles were collected by ultracentrifugation for 1 h at 125,000 g at 4°C. LmrA-enriched membrane vesicles were resuspended in 50 mM Hepes pH 7.0 with 150 mM NaCl and 10% glycerol, and 50 mM Tris-HCl pH 7.0 supplemented with 10% glycerol in the case of LmrCD was used. Samples were stored at −80°C in small aliquots.
Hoechst 33342 transport assay
The assay was basically performed as described in Lubelski et al. (Citation2006b). In brief, Hoechst 33342 becomes fluorescent upon partitioning into a hydrophobic environment, while it is essentially non-fluorescent in the aqueous phase. This allows monitoring the movement of the dye between the two phases (Shapiro and Ling Citation1995). For transport assays, membrane vesicles (0.5 mg of protein) from cells expressing wild-type LmrCD were incubated in 1 ml transport buffer (50 mM Tris-HCl pH 7.0, 2 mM MgSO4, 2 mM MgCl2 and 0.5 μM Hoechst 33342 with and without 30 μM of the assayed compound) at 30°C. The concentration of 30 μM for the tested compounds was chosen, because here the assay with the reference compound zosuquidar resulted in a significant change in Hoechst 33342 transport compared to the control measurement. The initial increase of fluorescence intensity indicates the partitioning of the drug into the membrane. After reaching equilibrium as indicated by a stable fluorescent signal, 5 μmol ATP was added to initiate transport out of the membrane, which was detected as a decrease of the fluorecence signal. The measurements were performed with a Fluorescence spectophotometer (Fluorolog II Horiba) with a magnetic stirred holder at 30°C using emission and excitation wavelengths of 355 and 457 nm, respectively. The transport activity was quantified as the slope of the linear part of the transport curve. The time window for the slope determination was 100 sec starting always 1 min after the addition of ATP.
Isolation of plasma membranes from Saccharomyces cerevisiae
Cells were grown in YPD at 30°C. At an OD600 of 1.5, a 10th volume of 5× YP (50 g/l yeast extract and 100 g/l tryptone/peptone) was added and cells were harvested at an OD600 of 3.5. The isolation of plasma membranes was performed as described (Kolaczkowski et al. Citation1996, Ernst et al. Citation2008). All steps of the preparation were performed at 4°C and all buffers contained a protease inhibitor mixture (complete EDTA-free; Roche) except the storage buffer. Cells were resuspended in 50 mM Tris-acetate (pH 8.0) and 5 mM EDTA and disrupted by vigorous shaking with glass beads. Unbroken cells and cell debris were removed by two centrifugation steps (2,000 g and 3,000 g for 5 min each). The resulting supernatant was centrifuged for 40 min at 20,000 g. Pellets were resuspended in 10 mM Tris-acetate (pH 7.5) and 0.2 mM EDTA and adjusted to a protein concentration of 5 mg/ml. Mitochondrial membranes were removed by acetic acid precipitation at pH 5.2 and subsequent centrifugation (6,900 g for 5 min). The supernatant was quickly adjusted to pH 7.5 and centrifuged for 30 min at 26,500 g. The pellets, plasma membranes, were resuspended in storage buffer (50 mM Hepes pH 7.0), and adjusted to a protein concentration of about 1 mg/ml. The quality of the samples was assessed by SDS-PAGE and rhodamine 6G transport activity.
Rhodamine 6G transport assay
The assay was performed as described (Ernst et al. Citation2008). In brief, a sample of plasma membranes (30 μg of protein) was diluted into 1 ml of transport buffer (50 mM Hepes, pH7.0, 10 mM MgCl2, and 150 nM rhodamine 6G) and incubated at 35°C. The excitation and emission wavelengths were set to 529 nm and 553 nm. Transport was initiated by the addition of ATP to a final concentration of 10 mM. The transport activity was determined as described for the Hoechst 33342 transport.
ATPase activity assay
The ATPase activity was monitored by the liberation of inorganic phosphate, which was quantified with molybdate/malachite green as described (Baykov et al. Citation1988). The reactions were performed in duplicate in a total volume of 200 μl in transport buffer or, in the case of LmrA, in 50 mM Hepes buffer supplemented with 150 mM NaCl and 5 mM MgCl2. For the assays between, 0.2 and 1 μg of protein was used, depending on the activity of the protein and 30 μM of a compound for the measurements of LmrA/CD and 5 μM for Pdr5. In order to determine the ATPase activity during transport, we added the transport substrates in the same concentrations as those used for the transport measurements (150 nM rhodamine 6G for Pdr5 and 500 nM Hoechst 33342) in the case of LmrCD and Pdr5. The reaction was started by the addition of 1 mM ATP and samples were incubated for 30 min at 30°C for LmrA and LmrCD and for 30 min at 35°C for Pdr5. After the addition of ATP, the reaction was stopped by transferring 25 μl of the sample into 175 μl 20 mM H2SO4. Subsequently 50 μl dye solution [0.096% (w/v) malachite green. 1.48% (w/v) ammonium molybdate, and 0.173% (w/v) Tween-20 in 2.36 M sulfuric acid] was added. After 15 min, the amount of free phosphate was quantified spectroscopically by the absorption at 620 nm. For the measurements, all appropriate controls were performed and subtracted where necessary. For calibration of free phosphate, Na2HPO4 was used as a standard. Control membrane vesicles for the measurements of LmrA were isolated from cells that expressed the ATPase inactive E512Q mutant of LmrA. For LmrCD membrane vesicles from cells that had only the empty expression vector, were used, while of Pdr5, the oligomycin-sensitive ATPase activity was measured.
Results
To develop inhibitors for ABC-type efflux pumps, three model systems, Pdr5 from S. cerevisiae as well as LmrA and LmrCD from L. lactis, have been studied. As a lead structure for the synthesis of potential inhibitors zosuquidar, a third-generation inhibitor of P-glycoprotein – the paradigm of MDR-ABC-transporters – has been used. Based on the structure-activity-relationship (Suzuki et al. Citation1997) for P-glycoprotein, a set of 26 zosuquidar derivatives were synthesized (Dittrich et al. Citation2012). To address the effects of the synthesized compounds, the ATPase, as well as transport activities for model substrates in the presence or absence of the potential inhibitors of Pdr5, LmrA and LmrCD, were studied.
Inhibition of Pdr5 activity by the synthesized zosuquidar derivatives
For the ABC-type efflux pump Pdr5, rhodamine 6G transport as well as its oligomycin-sensitive ATPase activity, were analyzed in plasma membrane vesicles (Ernst et al. Citation2008). These experiments revealed a substantial decrease (>50%) in the relative transport activity of the protein for many of the potential inhibitors (18 out of 27 compounds tested, see ). Similarly, the ATPase activity of Pdr5 was significantly reduced (>50%) by many of the compounds analyzed (10 out of 27 compounds tested). The reduction of ATPase activity was generally not as pronounced as the reduction of rhodamine 6G transport activity (see ).
Table I. ATPase and transport activity of Pdr5, LmrA and LmrCD in the presence of zosuquidar (1) or zosuquidar derivatives (2- 27); 100% activity corresponds to ATPase or transport activity of Pdr5, LmrA or LmrCD in the absence of zosuquidar or the derivatives. The assays were performed as described in Materials and method s. The ATPase activity of Pdr5 and LmrCD was determined in the presence of 150 nM rhodamine 6G and 500 nM Hoechst 33342, respectively. The reported values represent the average of three independent experiments with the standard deviation given as error.
In more detail, the transport data reveals that five compounds (6, 7, 25, 11 and 10, see ) reduce rhodamine 6G transport of Pdr5 significantly more (99–84% inhibition) than the lead compound zosuquidar (76% inhibition). Moreover, seven other compounds (2, 3, 4, 16, 22, 23 and 24) display a rhodamine 6G transport activity similar to zosuquidar (24%), while the other compounds show an intermediate (>33%) to low (>80%) inhibition of rhodamine 6G transport. Interestingly, the substituent in the para position of the aromatic rings seems to have only a minor influence on the inhibitory potency of compounds 2, 3 and 4 (24%, 24% and 22%). In contrast, the substituent in the para position of compounds 8, 9 and 10 has a drastic effect on rhodamine 6G transport. Fluorine atoms (compound 10) resulted in a strong inhibition (14%), while methyl groups (compound 9) had a drastically reduced effect (51%) and protons (compound 8) showed almost no effect (83%). Compounds 2, 3, 4 and 10 act as inhibitors with similar potency as zosuquidar (1), likely because their diarylmethyl structural unit is similar to the annulated benzene rings in the suberyl moiety of zosuquidar. Compound 25 contains a novel catechol acetal structure, which also provides substantial inhibitory activity and results in an almost 50% reduction of the rhodamine 6G transport relative to zosuquidar (13% vs. 24%). This result is unexpected and surprising in view of the severe geometrical differences of the compounds 2, 3, 4 and 10 and compound 25. In addition to compound 25, also the acetals 22, 23 and 24 result in a substantial inhibition of rhodamine 6G transport (31, 27 and 31%), a magnitude, which is similar to zosuquidar.
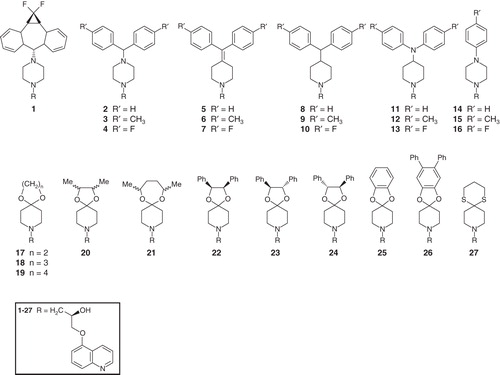
To address the inhibitory potency of the above-mentioned compounds in more detail, selected substances representative for the different classes of the zosuquidar derivatives were analyzed with regard to their IC50 value for transport and ATPase activity. The analysis of compounds 2, 7, 11 and 23 (see ) resulted in IC50 values for rhodamine 6G transport in the high nanomolar range (∼ 300–940 nM). Surprisingly, the IC50 value of compound 11 was significantly lower than the one of compound 7 (300 nM vs. 940 nM), although in our initial experiments, compound 7 inhibited rhodamine 6G transport of Pdr5 to a much higher degree than 11 (1% vs. 14%). The reason for this difference is, however, not a different inhibition potential of the synthesized compounds as all compounds were able to completely inhibit rhodamine 6G transport (see ). Furthermore, our analysis revealed IC50 values for the ATPase activity in the low micromolar range [∼ 4 (compound 23) to 20 μM (compound 2)]. Thus, the synthesized zosuquidar derivatives seem to impair rhodamine 6G transport at much lower concentrations compared to the ATPase activity. Furthermore, among the zosuquidar derivatives effectively interfering with Pdr5 activity very different structures of the hydrophobic moiety are evident, which like compound 7 and 25, differ substantially in their structure with the phenyl rings in plane with the piperidine skeleton in compound 7, while a perpendicular orientation is observed in compound 25. This observation suggests that the binding site of Pdr5 is relatively ‘tolerant' with regard to alterations in the hydrophobic moiety of zosuquidar, in line with the results of earlier SAR studies on P-glycoprotein (Starling et al. Citation1997, Suzuki et al. Citation1997).
Figure 2. Determination of IC50 values for rhodamine 6G transport (A–D) and ATPase activity (E–H) of Pdr5. The activity of Pdr5 was determined with increasing concentrations of 2 (A & E), 7 (B & F), 11 (C & G) and 23 (D & H). The transport and ATP assays were performed in the presence of the indicated compounds as described in Materials and methods.
Effect of the synthesized potential inhibitors on LmrA
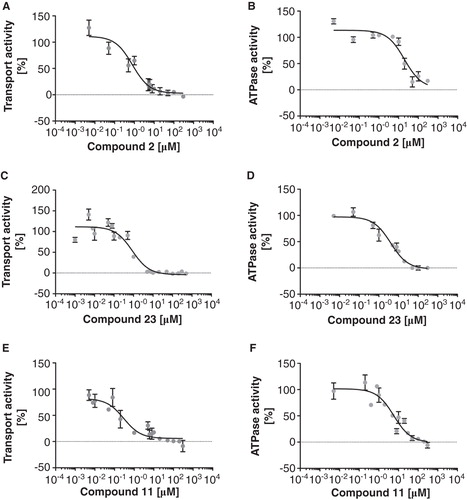
To address the potency of the synthesized compounds on LmrA as one model system for MDR efflux pumps of Gram-positive bacteria, their effect on ethidium efflux of L. lactis cells overexpressing LmrA was studied (). Interestingly, the re-energetization of ATP-depleted and ethidium-loaded L. lactis cells expressing wild-type LmrA resulted in a drastic increase of ethidium fluorescence while the ATPase-deficient E512Q mutant (van den Berg van Saparoea et al. Citation2005, although Venter et al. Citation2008 reports different values) of LmrA and control experiments with ATP-depleted cells (addition of water instead of glucose) resulted in a relatively small increase of fluorescence (see ). Furthermore, the expression of the LmrCD (Lubelski et al. Citation2004) results in a similar increase of ethidium fluorescence upon addition of glucose (). Thus, our data suggest that the extrusion of ethidium from L. lactis cells by LmrA (or LmrCD) can be monitored by an increase in ethidium fluorescence. The latter is likely due to the extrusion of free dye from the cell, because free ethidium quenches the fluorescence of the DNA bound ethidium, which is highly fluorescent (Heller and Greenstock Citation1994). Since this quenching is reduced by the extrusion of free dye from the cell, this results in a de-quenching of fluorescence and thus in an increase of the overall ethidium fluorescence. Moreover, our data is supported by fluorescence microscopy of L. lactis cells overexpressing LmrA. Here, the fluorescence of ethidium in ATP-depleted cells is drastically reduced upon addition of glucose in L. lactis expressing wild-type LmrA, but not in cells expressing the ATPase-deficient E512Q mutant (see ).
Figure 3. Ethidium transport by L. lactis cells expressing LmrA, LmrA E512Q or LmrCD. Green line: LmrA after the addition of glucose; dashed, green line: LmrA after the addition of water as control; dotted, green line: LmrA after the addition of glucose and compound 1; black line: LmrA E512Q after the addition of glucose; blue line: LmrCD after the addition of glucose. An arrow indicates the addition of glucose, water or glucose and compound 1. The transport assay was performed as described in Materials and methods. This Figure is reproduced in colour in Molecular Membrane Biology online.
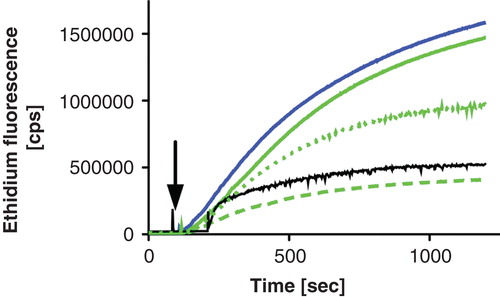
Figure 4. Confocal LSM images of ATPase-deficient LmrA E512Q expressing L. lactis cells in the absence (A) or presence (B) of glucose and of LmrA expressing L. lactis cells in the absence (C) or presence (D) of glucose. Cells were loaded with 40 μM ethidium and pictures were acquired as outlined in Materials and methods. This Figure is reproduced in colour in Molecular Membrane Biology online.
It has been reported that the expression of LmrA in L. lactis results in a decrease of ethidium fluorescence upon energization of the cells by the addition of glucose, which was not observed for control cells not expressing LmrA (Balakrishnan et al. Citation2004). Nevertheless, comparison of the assays reveals substantial differences among them, which might account for the different observation. Most strikingly, the assays used very different ethidium concentrations of 5 μM (Balakrishnan et al. Citation2004) vs. 40 μM (this study) and in addition, our assay includes a washing step to remove free ethidium, while Balakrishnan et al. (Citation2004) did not remove free dye. Thus, since free dye quenches the fluorescence of DNA-bound ethidium (Heller and Greenstock Citation1994), it seems plausible that our assay, due to the high concentration of ethidium, monitors the extrusion of free dye (i.e., an increase of ethidium fluorescence) while Balakrishnan et al. (Citation2004) analyzed the extrusion of DNA bound ethidium (i.e., a decrease in ethidium fluorescence).
As already pointed out, the effect of the synthesized compounds on the activity of LmrA was analyzed by their impact on whole cell ethidium transport of the protein as well as its ATPase activity in membrane vesicles. As expected, the compounds show very diverse effects on the ethidium efflux of LmrA with some compounds showing hardly an effect such as compound 2 (100%) while others like compound 8 reduce the ethidium efflux substantially (30% residual transport activity compared to the control) and only few compounds (for example compound 16) stimulate transport activity of LmrA (148%). In contrast, the ATPase activity of LmrA is stimulated by many compounds (for example compound 15, 143%), while a substantial inhibition of ATPase activity (for example compound 6, 79%) is only observed for a few compounds (see ). In detail our data on the ethidium efflux reveal that seven of the 26 compounds tested (27, 22, 23, 7, 11, 10 and 8; see ) resulted in a higher inhibition of ethidium efflux (30–48%) than zosuquidar (52%). Interestingly, the acetals 22 and 23 substantially impaired ethidium efflux of LmrA (35% and 41%) while their diastereomer 24 had basically no effect (95%) on transport. Moreover acetal 25, which was even more efficient than 22 and 23 in the case of Pdr5, hardly showed an effect on ethidium efflux of LmrA. Furthermore, in compounds with a diarylmethyl structural unit containing a fluorine atom (for example compound 7) or a proton (for example compound 8) in the para position of the aromatic rings resulted in compounds that significantly impaired ethidium efflux of LmrA in several cases, while an effect like this was never observed for methyl groups. Therefore, it is not straightforward to deduce a rational structure-activity-relationship for ethidium transport of LmrA. Furthermore, the synthesized compounds had hardly an inhibitory effect on the ATPase activity of LmrA. Rather, many substances stimulated the ATPase activity of the protein.
Inhibition of Hoechst 33342 transport and ATPase activity of LmrCD
LmrCD from L. lactis was chosen as a second model system due to its significant sequence homology (50–60% amino acid identity) with MDR-ABC-transporters of clinically relevant Gram-positive bacteria (e.g., Streptococcus pneumoniae) (Lubelski et al. Citation2007). To address the activity of LmrCD, a published Hoechst 33342 transport assay (Lubelski et al. Citation2006b) in combination with an analysis of the ATPase activity in plasma membranes was employed. In contrast to the previously described efflux pumps Pdr5 and LmrA, our data on Hoechst 33342 transport for LmrCD reveal a very specific inhibitory effect with only three compounds (2, 12 and 24, see ) showing a potent inhibition of Hoechst 33342 transport (28, 20 and 11% compared to the control), while all other compounds including zosuquidar (42%) displayed only a reduced inhibitory activity towards the protein (see ). Interestingly for compounds 2 and 12, relatively small modifications in the substituent of the aromatic rings of the hydrophobic moiety had quite drastic effects on the inhibitory potency. For example, the addition of a fluorine atom or a methyl group in the para position of the aromatic rings of compound 2 results in an increase of the relative transport activity of LmrCD from 29–65%. Similar consequences were observed for compound 12 where the removal of the methyl group in the para position of the aromatic rings or its replacement by a fluorine atom resulted in an increase of the relative transport activity of the protein from 20–77% and 73%, respectively. Nevertheless, the most intriguing result of our studies on LmrCD is the fact that compound 24 reduces the relative transport activity of the protein to 11%. In other words, compound 24 is almost four times as potent as zosuquidar, while its diastereomers 22 and 23 have basically no effect (97% and 96%). In contrast to the transport activity, ATPase activity was not (compounds 2 and 24) or only moderately (compound 12; 62%) affected. Thus, LmrCD can be specifically inhibited by only three of the synthesized derivatives of zosuquidar. This is amazing keeping the very similar chemical structures of the compounds 2 (inhibition) and 13 (no inhibition) and the stereo-selectivity with compounds 22, 23 (no inhibition) and 24 (inhibition) in mind.
Discussion
Inhibition of model systems for ABC-type efflux pumps of pathogenic microorganisms
As described in the Results section, our study revealed very different effects of the synthesized compounds on the systems analyzed. For Pdr5, a model system for ABC efflux pumps from pathogenic fungi, 12 out of 26 of the synthesized compounds resulted in a significant reduction of rhodamine 6G transport with an inhibition of the protein similar to or better than that of the lead compound zosuquidar. In addition, determination of the IC50 values for transport for selected zosuquidar derivatives (compounds 2, 7, 11 and 23) revealed high nanomolar affinities. Unfortunately, it is difficult to derive a reasonable structure-activity relationship from our results on Pdr5 to guide further improvement of the inhibitory potency of these compounds. Nevertheless, in comparison to inhibitors of Pdr5 identified so far, our compounds are –with one exception – among the most potent ones. So far only the hydrophobic estradiol-derivative RU49953 with an IC50 value of 0.083 μM had a significantly higher inhibitory potency while other compounds like the estradiol-derivative RU38486 (0.54 μM), prenyl-flavonoids 6-(3,3-DMA)galangin (0.24 μM) and 6-geranylchrysin (0.43 μM) as well as paclitaxel (2.8 μM) or verapamil (1.7 μM) had a similar or reduced inhibitory potency towards rhodamine 6G transport of Pdr5 (Conseil et al. Citation2000, Citation2003).
Similar to Pdr5, the ethidium efflux of LmrA could be substantially inhibited by seven of the compounds synthesized, but a reasonable structure-activity relationship could not be established. Comparison of the most potent inhibitors of transport for Pdr5 and LmrA reveals a substantial overlap among the inhibitors of these proteins (see ), while no such overlap exists for LmrCD with either Pdr5 or LmrA. Nevertheless, one has to keep in mind that three different substrates were used in the transport assays, which complicates the comparison of the results. For P-glycoprotein, the prototype of a MDR ABC-transporter, different substrate-binding pockets have been identified in the substrate-binding site each responsible for binding a certain subset of substrates (Shapiro and Ling Citation1997). Therefore, one explanation could be that the pockets binding the different transport substrates might preferentially interact with different types of inhibitors. This will obviously complicate any rational approach to improve specificity of these inhibitors. However, modifications of ‘only' the hydrophobic wieso geht doch für jedes Protein einzeln moiety of zosuquidar, which in the case of the P-glycoprotein proved to be of pivotal importance for the inhibitory potency (Suzuki et al. Citation1997), lead to the identification of inhibitors with nanomolar affinities for Pdr5. This of course might suggest new lead structures for drug development against fungal ABC transporters.
Figure 5. Schematic summary of the zosuquidar derivatives that displayed the most prominent effect on transport of LmrA, LmrCD and Pdr5. Substances were grouped by circles or a box according to the ABC transporter for which they showed the most prominent effect to highlight the partial overlap between the substrate binding sites of LmrA and Pdr5.
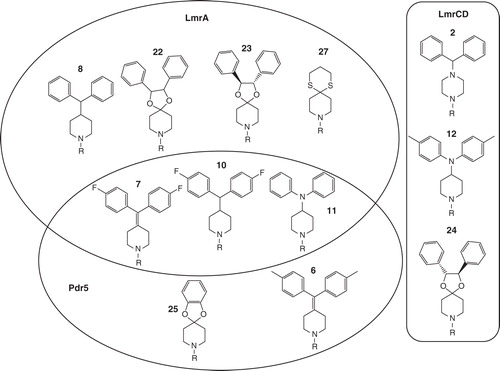
In contrast to Pdr5 and LmrA, only three of the compounds tested significantly reduced the Hoechst 33342 transport of LmrCD. Furthermore, modifications in the diarylmethyl unit of compounds 2 and 12 resulted in a substantial reduction of inhibitory potency, with the Hoechst transport of LmrCD increasing, for example, from 28% for compound 2 to 64% and 65% upon substitution of the proton in the para position of the aromatic ring of compound 2 by either a methyl group (compound 3) or a fluorine atom (compound 4). Moreover, in the case of compound 24, even the stereochemistry of the compound is important, because its diastereomers 22 and 23 have basically no effect on the transport activity of LmrCD. Therefore, compounds 2, 12 and especially 24 seem to be very promising lead structures for further improving the inhibitory potency towards LmrCD, which in view of its homology with efflux pumps of pathogenic Gram-positive bacteria, might be of considerable interest for drug development.
Conclusions
We have identified potent inhibitors of the ABC-type efflux pumps Pdr5, LmrA and LmrCD. For Pdr5, inhibitors with high nanomolar affinities have been discovered. The inhibitors found are, with the exception of RU49953, among the most potent inhibitors known for Pdr5. One of the compounds identified, catechol acetal 25, has the potential to become the lead compound of a new class of inhibitors as similar structures have not been reported before. In contrast to Pdr5, until now, no inhibitors have been reported for LmrCD, homologs of which have been implicated in multidrug resistance of Gram-positive pathogens. Thus, our study is – to the best of our knowledge – the first report of potent inhibitors of this protein. Among the inhibitors identified especially acetal 24 is highly interesting due to its high stereospecificity.
Acknowledgements
We thank Stefanie Weidtkamp-Peters for excellent support during the microscopy experiments. We are indebted to Petra Küppers and Patrick Bakkes for support during various stages of the project and Rakeskumar Gupta for the Pdr5 samples. This work was support by the VW Foundation (I82/605) and the EU (EDICT (European Drug Initiative on Channels and Transporters) FP7 Theme [Health-2007-2.1.1.-5]) to L.S.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Balakrishnan L, Venter H, Shilling RA, Van Veen HW. 2004. Reversible transport by the ATP-binding cassette multidrug export pump LmrA: ATP synthesis at the expense of downhill ethidium uptake. J Biol Chem 279:11273–11280.
- Baykov AA, Evtushenko OA, Avaeva SM. 1988. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem 171:266–270.
- Conseil G, Decottignies A, Jault JM, Comte G, Barron D, Goffeau A, 2000. Prenyl-flavonoids as potent inhibitors of the Pdr5p multidrug ABC transporter from Saccharomyces cerevisiae. Biochemistry 39:6910–6917.
- Conseil G, Perez-Victoria JM, Renoir JM, Goffeau A, Di Pietro A. 2003. Potent competitive inhibition of drug binding to the Saccharomyces cerevisiae ABC exporter Pdr5p by the hydrophobic estradiol-derivative RU49953. Biochim Biophys Acta 1614:131–134.
- Dantzig AH, Law KL, Cao J, Starling JJ. 2001. Reversal of multidrug resistance by the P-glycoprotein modulator, LY335979, from the bench to the clinic. Curr Med Chem 8:39–50.
- Dittrich T, Hanekop N, Infed N, Schmitt L, Braun M. 2012. Synthesis of 5-oxyquinoline derivatives for reversal multidrug resistance. Beilstein J. Org. Chem 8:1700–1704.
- Egner R, Rosenthal FE, Kralli A, Sanglard D, Kuchler K. 1998. Genetic separation of FK506 susceptibility and drug transport in the yeast Pdr5 ATP-binding cassette multidrug resistance transporter. Mol Biol Cell 9:523–543.
- Ernst R, Kueppers P, Klein CM, Schwarzmueller T, Kuchler K, Schmitt L. 2008. A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug ABC transporter Pdr5. Proc Natl Acad Sci USA 105:5069–5074.
- Fukazawa N, Odate M, Suzuki T, Otsuka K, Tsuruo T, Sato W. 1990. Preparation of heterocyclic compounds as anticancer drug potentiators. EP 363212.
- Fukazawa N, Suzuki T, Otsuka K, Yano O, Sato W, Tsuruo T. 1992. New quinolines, their preparations, and anticancer activity enhancers containing the quinolines. Japanese patent 04235983.
- Golin J, Ambudkar SV, Gottesman MM, Habib AD, Sczepanski J, Ziccardi W, 2003. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J Biol Chem 278:5963–5869.
- Gottesman MM, Fojo T, Bates SE. 2002. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat Rev Cancer 2:48–58.
- Heller DP, Greenstock CL. 1994. Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye. Biophys Chem 50:305–312.
- Kolaczkowski M, Van Der Rest M, Cybularz-Kolaczkowska A, Soumillion JP, Konings WN, Goffeau A. 1996. Anticancer drugs, ionophoric peptides, and steroids as substrates of the yeast multidrug transporter Pdr5p. J Biol Chem 271:31543–31548.
- Lubelski J, De Jong A, Van Merkerk R, Agustiandari H, Kuipers OP, Kok J, 2006a. LmrCD is a major multidrug resistance transporter in Lactococcus lactis. Mol Microbiol 61:771–781.
- Lubelski J, Konings WN, Driessen AJ. 2007. Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiol Mol Biol Rev 71:463–476.
- Lubelski J, Mazurkiewicz P, Van Merkerk R, Konings WN, Driessen AJ. 2004. ydaG and ydbA of Lactococcus lactis encode a heterodimeric ATP-binding cassette-type multidrug transporter. J Biol Chem 279:34449–34455.
- Lubelski J, Van Merkerk R, Konings WN, Driessen AJ. 2006b. Nucleotide-binding sites of the heterodimeric LmrCD ABC-multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry 45:648–656.
- Morschhauser J. 2010. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet Biol 47:94–106.
- Pfister JR, Lynne SD. 1994. 10,11-Methanodibenzosuberane derivatives used as chemosensitizing agents.
- Pfister JR, Makra F, Muehldorf AV, Wu H, Nelson JT, Cheung P, 1995. Methanodibenzosuberylpiperazines as potent multidrug-resistance reversal agents. Bioorgan Medic Chem Lett 5:2473–2476.
- Piddock LJ. 2006a. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev 19:382–402.
- Piddock LJ. 2006b. Multidrug-resistance efflux pumps – not just for resistance. Nat Rev Microbiol 4:629–636.
- Prasad R, De Wergifosse P, Goffeau A, Balzi E. 1995. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr Genet 27:320–329.
- Sanglard D, Coste A, Ferrari S. 2009. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res 9:1029–1050.
- Shapiro AB, Ling V. 1995. Reconstitution of drug transport by purified P-glycoprotein. J Biol Chem 270:16167–16175.
- Shapiro AB, Ling V. 1997. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem 250:130–137.
- Sircar I, Haleen SJ, Burke SE, Barth H. 1992. Synthesis and biological activity of 4-(diphenylmethyl)-alpha-[(4-quinolinyloxy)methyl]-1-piperazineethanol and related compounds. J Med Chem 35:4442–4449.
- Sorbera LA, Castaner J, Silvestre JS, Bayes M. 2003. Zosuquidar trihydrochloride – multidrug resistance modulator P-glycoprotein (MDR-1) inhibitor. Drugs Future 28:125–136.
- Starling JJ, Shepard RL, Cao J, Law KL, Norman BH, Kroin JS, 1997. Pharmacological characterization of LY335979: A potent cyclopropyldibenzosuberane modulator of P-glycoprotein. Adv Enzyme Regul 37:335–347.
- Suzuki T, Fukazawa N, San-Nohe K, Sato W, Yano O, Tsuruo T. 1997. Structure-activity relationship of newly synthesized quinoline derivatives for reversal of multidrug resistance in cancer. J Med Chem 40:2047–2052.
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. 2006. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234.
- Van Den Berg van Saparoea HB, Lubelski J, Van Merkerk R, Mazurkiewicz PS, Driessen AJ. 2005. Proton motive force-dependent Hoechst 33342 transport by the ABC transporter LmrA of Lactococcus lactis. Biochemistry 44:16931–16938.
- Van Veen HW, Callaghan R, Soceneantu L, Sardini A, Konings WN, Higgins CF. 1998. A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature 391:291–295.
- Van Veen HW, Margolles A, Muller M, Higgins CF, Konings WN. 2000. The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J 19:2503–2514.
- Van Veen HW, Venema K, Bolhuis H, Oussenko I, Kok J, Poolman B, 1996. Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc Natl Acad Sci USA 93:10668–10672.
- Venter H, Velamakanni S, Balakrishnan L, Van Veen HW. 2008. On the energy-dependence of Hoechst 33342 transport by the ABC transporter LmrA. Biochem Pharmacol 75:866–874.