Abstract
Human lipocalin-1 interacting membrane receptor (LIMR) was the first lipocalin receptor to be identified, as a specific receptor for lipocalin-1 (Lcn1). Subsequently LIMR has been reported to interact with other ligands as well, notably with the bovine lipocalin β-lactoglobulin (BLG) and with the unrelated secretoglobin uteroglobin (UG). To study the ligand-binding behaviour of this prototypic lipocalin receptor in more detail, a system was developed for the recombinant expression of LIMR in Drosophila Schneider 2 (S2) cells, and for the subsequent solubilization and purification of the protein. The receptor forms dimers or larger oligomers when solubilized in n-dodecyl β-D-maltoside (DDM). The full-length, functional receptor was captured onto a surface plasmon resonance (SPR) chip via an α-V5 antibody, and the binding of various potential ligands was followed in time. In this way, LIMR was shown to be highly specific for Lcn1, binding the lipocalin with low micromolar to high nanomolar affinity. No interactions with any of the other putative ligands could be detected, raising doubts about the physiological relevance of the reported binding of BLG and UG to the receptor.
Introduction
The lipocalins are a family of small, soluble, secreted proteins, characterized by their typical fold of an antiparallel eight-stranded β-barrel, with an α-helix attached (Akerstrom et al. Citation2000, Citation2006, Flower et al. Citation2000). They bind and transport mainly hydrophobic molecules, and play a role in a wide range of biological processes, such as animal behaviour (Briand et al. Citation2004), immunity (Flo et al. Citation2004), iron metabolism (Devireddy et al. Citation2005), signal transduction (Yang et al. Citation2005), development (Clagett-Dame and DeLuca Citation2002) and vision (Quadro et al. Citation1999). Although the lipocalins themselves have been studied extensively, both structurally (Flower Citation2000b, Akerstrom et al. Citation2006) and functionally (Akerstrom et al. Citation2000, Citation2006), little is known about the specific membrane receptors to which many lipocalins bind (Flower Citation2000a, Burke et al. Citation2005, Akerstrom et al. Citation2006).
To date, four such receptors have been described. Megalin is a high-capacity, low-affinity, wide-specificity endocytic receptor, which binds many ligands to its large extracellular domain, including several lipocalins (Christensen and Birn Citation2002). The first specific lipocalin receptor to be identified was lipocalin-1 interacting membrane receptor (LIMR) (Wojnar et al. Citation2001). More recently, a receptor for the mouse lipocalin 24p3 was cloned, named 24p3R, which internalizes the lipocalin and thereby regulates intracellular iron levels and apoptosis (Devireddy et al. Citation2005). Finally, the receptor for retinol binding protein (RBP) has been identified as STRA6, and confirmed to bind this lipocalin with high specificity and affinity and to mediate the uptake of retinol from holo-RBP (Sivaprasadarao and Findlay Citation1988a, Citation1988b, Kawaguchi et al. Citation2007). There is no sequence homology amongst this group of receptors and very little with other receptor families. However, there might be similarities in their topography in the bilayer.
LIMR is a 55-kDa membrane protein with nine putative transmembrane segments, an extracellular N-terminus that presumably forms part of the lipocalin binding site, a large intracellular loop and intracellular C-terminus (Wojnar et al. Citation2001). It is widely expressed in human tissues (Wojnar et al. Citation2001), and has orthologs in many other species (Wojnar et al. Citation2003). Although it was originally identified as a specific receptor for lipocalin-1 (also termed tear lipocalin or von Ebner's gland protein, here abbreviated, Lcn1), through screening of a cDNA phage-display library of human pituitary gland with this lipocalin as bait (Wojnar et al. Citation2001), subsequently interactions of the receptor with Lcn1's porcine homolog von Ebner's gland protein (pVEG) (Burke Citation2004), the secretoglobin uteroglobin (UG) (Zhang et al. Citation2006) and the bovine lipocalin from milk β-lactoglobulin (BLG) (Fluckinger et al. Citation2008) have also been reported.
Lcn1 is a highly promiscuous lipocalin (Glasgow et al. Citation1995), with an unusually large cavity (Breustedt et al. Citation2005), which accommodates many ligands with diverse structures and chemical properties, e.g. fatty acids and alcohols, lipids, steroids, siderophores (Fluckinger et al. Citation2004) and lipid oxidation products (Lechner et al. Citation2001). It is expressed mainly at epithelial surfaces, where it is presumed to have a scavenging function, removing potentially harmful xenobiotics (Gasymov et al. Citation2005). Furthermore, it comprises 20–30% of the protein fraction of human tear fluid (Fullard and Tucker Citation1991), where it reduces the surface tension (Millar et al. Citation2009), and contributes to tear viscosity (Gouveia and Tiffany Citation2005), by binding lipids and interacting with other proteins when in the holo-state. It was seen to interact in vitro with a short N-terminal fragment of LIMR (Wojnar et al. Citation2001), and to be internalized by human teratocarcinoma NT2 cells and human T-47D breast cancer cells in an LIMR-dependent manner (Wojnar et al. Citation2003).
UG is a multifunctional secreted protein, which was shown, via radioligand-binding assays, to bind with high specificity and affinity (K d = 18 nM) to COS-1 and HTB-81 cells recombinantly expressing LIMR (Zhang et al. Citation2006). The suggestion is that UG suppresses cell motility and invasion via this receptor (Zhang et al. Citation2006). Although UG shares some basic characteristics with the lipocalins, such as binding of retinoids and other lipophilic compounds, and a role in the immune system, their structures are quite different (Morize et al. Citation1987, Breustedt et al. Citation2005).
BLG, the major milk whey protein of most mammals, but not humans (Kontopidis et al. Citation2004), is similar in sequence (Redl Citation2000) and structure (Papiz et al. Citation1986, Breustedt et al. Citation2005) to Lcn1. This lipocalin is able to bind a wide range of ligands (Kontopidis et al. Citation2002), though its cavity is somewhat smaller as compared to Lcn1. It was shown to copurify with a short N-terminal fragment of LIMR, and to be internalized by NT2 and Sf9 cells in an LIMR-dependent manner (Fluckinger et al. Citation2008).
It is unclear how LIMR, which is predicted to have a relatively small extracellular domain (Wojnar et al. Citation2001), can bind such different ligands, and what the biological significance is, especially of the described interactions with non-human proteins. To further the understanding of this lipocalin receptor, the solubilization, purification and initial characterization of the protein is described here for the first time. We studied the interactions of LIMR with all the proposed ligands, both qualitatively and quantitatively, using mainly surface plasmon resonance (SPR). Our data suggest that LIMR only has high affinity (high nanomolar to low micromolar) for Lcn1.
Methods
Plasmids
The human LIMR coding sequence was amplified by PCR from the vector pFastBac/LIMR using primers 5′-ACC TGA AGA TCT GTC GAC GCA ATG GAA GCA CCT GAC TAC GAA GTG CTA-3′ and 5′-CCC TCT AGA AAG CTT CTG GTG CTG GGT C-3′. The amplification product was subcloned into the Drosophila expression vector pMT/BiP/V5-His A (Invitrogen), in frame with the N-terminal BiP secretion signal to direct LIMR towards the plasma membrane and with the C-terminal V5- and 6His-tags, using restriction enzymes BglII and XbaI.
The coding sequence for human Lcn1 (comprising amino acids 19–176, without the N-terminal signal sequence for secretion) was obtained (Burke Citation2004) in the bacterial expression vector pQE-70 (Qiagen). The coding sequence for its porcine homolog pVEG (comprising amino acids 20–176, without the N-terminal signal sequence for secretion) was amplified by PCR from pQE-30/pVEG using primers 5′-CGT CAC CAT GGC GCA GGA GTT CCC GGC CGT G-3′ and 5′-GCA TCG CGG CCG CGC GGC CTT CAA TGT TCC CTC CTG GAG AGC A-3′. The amplification product was subcloned into the expression vector pQE-TriSystem (Qiagen), in frame with the C-terminal Strep- and 6His-tags, using restriction enzymes NcoI and NotI. The integrity of all constructs was confirmed by DNA sequencing.
Recombinant protein expression
Drosophila Schneider 2 (S2) cells were cultured at 28°C in Schneider's Drosophila medium (Invitrogen) supplemented with 10% (v/v) heat inactivated foetal bovine serum (FBS). Cells were maintained in loosely adherent or suspension culture. S2 cells were cotransfected with pMT/BiP/V5-His/LIMR and pCoHygro using the calcium phosphate transfection procedure (Invitrogen) according to the manufacturer's recommendations. Stably transfected cell lines were selected and maintained by addition of hygromycin B (Invitrogen) to the medium to a final concentration of 250 μg/ml. Protein expression was induced by adding copper sulphate to the medium to a final concentration of 500 μM. Cells were harvested one to five days after induction.
The lipocalins Lcn1 and pVEG were expressed in Escherichia coli BL21-CodonPlus(DE3)-RP and Rosetta-Gami 2, respectively. Bacterial cells, transformed with either pQE-70/Lcn1 or pQE-TriSystem/pVEG, were grown overnight in LB medium with the appropriate antibiotics, diluted 1:100 in fresh medium and incubated at 37°C with shaking (200 rpm) until the OD600 reached 0.6–0.8. Protein expression was induced by adding IPTG to the medium to a final concentration of 1 mM, and cells were collected 4 h after induction.
Protein purification
To prepare membranes from S2 cells, the cell pellet was suspended in phosphate buffer (20 mM NaH2PO4, pH 7.4, 0.15 M NaCl, 4 mM CaCl2, protease inhibitors [complete EDTA-free, Roche]), and subjected to five cycles of freezing the suspension in a dry ice/ethanol bath and thawing in a 37°C water bath to lyse the cells. The membrane fraction was separated from the soluble cell components by ultracentrifugation (100,000 g at 4°C for 1 h). Membranes were resuspended in phosphate buffer (as above) at a concentration of 10–20 mg/ml total protein. Vesicles were prepared by passing the mixture through a 25 G needle multiple times, always keeping the membranes on ice, until a homogeneous vesicle suspension was obtained.
LIMR was solubilized from S2 membranes using the mild, non-ionic detergent n-dodecyl β-D-maltoside (DDM, 1% [w/v]) and NaCl (0.5 M) in phosphate buffer (as above). The mixture was kept on ice for 90 min, while regularly passing it through a 25 G needle to aid solubilization. Insoluble material was subsequently removed by a 20 min spin at 100,000 g at 4°C.
LIMR was purified in two steps, utilising both the 6His- and the V5-tags on the protein. For the first step, the supernatant from the S2 membrane solubilization was supplemented with 20 mM imidazole and 10% (v/v) glycerol and incubated overnight at 4°C with Ni Sepharose 6 Fast Flow (GE Healthcare). The resin with bound LIMR was packed into a column, washed with 10 column volumes of buffer (20 mM NaH2PO4, pH 7.4, 0.5 M NaCl, 0.5% DDM, 10% glycerol, 40 mM imidazole) and eluted in the same buffer supplemented with 250 mM imidazole. The receptor was kept below 4°C. The eluate from the Ni Sepharose resin was subsequently incubated with α-V5-agarose (Sigma), for at least 90 min at 4°C with gentle agitation. The resin was loaded onto a column and washed with 10 column volumes of buffer (20 mM NaH2PO4, pH 7.4, 0.15 M NaCl, 0.5% DDM). LIMR was eluted by disrupting the antibody-antigen bonds in low pH buffer (0.1 M glycine, pH 2.5, 0.5% DDM). The eluate was collected directly into tubes containing neutralizing buffer (1 M Tris, pH 9). LIMR fractions were dialyzed against the appropriate buffer for the downstream experiment using the Slide-A-Lyzer MINI Dialysis system (Pierce, 10 kDa MWCO).
For lipocalin purification, bacterial cell pellets were resuspended in PBS (10 mM NaH2PO4, pH 7.4, 0.14 M NaCl, 3 mM KCl, protease inhibitors), and incubated with lysozyme (0.4 mg/ml) for 20 min at 37°C with shaking. After a further 30 min incubation with DNase I (2 U/ml), the suspension was centrifuged for 30 min at 4°C at 5240 g to remove all insoluble material.
The supernatant containing the lipocalin was purified on Ni Sepharose as described above for LIMR, except for the exclusion of detergent from all buffers (see Supplementary Figure S1, available online).
β-Lactoglobulin (Sigma) and uteroglobin (Abnova) were commercially acquired and used without further purification.
Protein analysis
To analyze LIMR expression and purification, samples were separated on SDS-PAGE gels (10% (w/v) acrylamide), and proteins were either directly visualized by silver staining (O’Connell and Stults Citation1997), or transferred to PVDF membrane and detected by Western blotting using α-V5-HRP antibody (Invitrogen) and enhanced chemiluminescence Western blotting substrate (Pierce). Silver stained bands were further analyzed by MALDI-MS peptide mass fingerprinting (PMF) (Breton et al. Citation2008).
Lipocalin samples were separated on SDS-PAGE gels (15% acrylamide), and proteins were visualized by Coomassie Blue staining. Protein concentrations were determined using the BCA assay (Smith et al. Citation1985).
Gel filtration
To determine the oligomeric state of DDM-solubilized LIMR, a concentrated sample of purified receptor was run over a Superose 6 column (GE Healthcare) in mild non-denaturing buffer (50 mM NaH2PO4, pH 7.4, 150 mM NaCl, 0.1% DDM, filtered and degassed). Fractions (60 of 0.5 ml each) were collected and analysed for the presence of LIMR by SDS-PAGE and Western blotting. The elution profile was compared with a standard curve calculated using the elution volumes of proteins from the Gel Filtration HMW Calibration Kit (GE Healthcare).
Surface plasmon resonance
To follow interactions between solubilized, purified LIMR and several of its ligands in real time, SPR experiments were performed using the Biacore 3000. CM5 sensor chips were used to immobilize α-V5 antibody by amine coupling chemistry, injecting first a 7-min pulse of a 1:1 mixture of 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and 0.1 M N-hydroxysuccinimide (NHS) to activate the surface, followed by the α-V5 antibody (10 μg/ml, Invitrogen) in pre-concentration buffer (10 mM acetate, pH 5.5) up until the desired level of immobilization (typically 5,000 resonance units [RU]). Finally a 7-min pulse of 1 M ethanolamine was administered to deactivate excess reactive groups. One flow cell per chip was activated and deactivated without immobilization of a protein, to use as an empty reference flow cell. As a running buffer HBS-EP (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% (v/v) surfactant P20, filtered and degassed) was used; 0.05% DDM was included in the running buffer when working with LIMR. All proteins were purified as described above and dialysed against running buffer. Flow rates were 5–10 μl/min for immobilization, capturing and qualitative binding analysis, 30–60 μl/min for quantitative kinetics experiments. SPR experiments were carried out at 25°C. Kinetic parameters for the binding of lipocalins and other putative ligands to LIMR were measured using single cycle kinetics (SCK) (Karlsson et al. Citation2006) combined with a receptor-on-chip set-up, an approach which has the advantage that surface regeneration is not required, since the same surface is used for the injection of five concentrations of a putative binding partner, each concentration a factor two or three higher than the last, followed by a long dissociation phase. BIA evaluation software was used to correct the signal for refractive index changes due to variations in buffer composition using the reference flow cell, and for drift caused by loss of receptor from the surface using five injections of buffer in a similar cycle. These doubly corrected data were then fitted using a model assuming a 1:1 interaction, which derives a value for k on from the association phase of the five different injections, and a value for k off from the final long dissociation phase.
Results
Expression of LIMR in Drosophila S2 cells
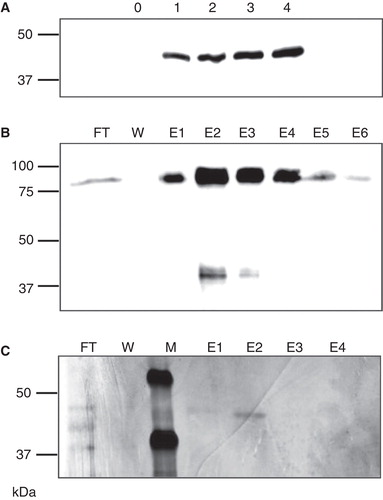
Drosophila S2 cells, stably transfected with vector pMT/BiP/V5-His/LIMR, express a V5-labelled product with an apparent molecular mass of ca. 42 kDa one to four days after induction (). The same product is expressed in transiently transfected S2 cells, from vectors with either a constitutive (pAc5.1/V5-His/LIMR) or an inducible (pMT/BiP/V5-His/LIMR) promoter, in mammalian HEK293 cells transfected with pcDNA3/LIMR, and in cell-free in vitro expression systems, but not in non-transfected or non-induced cells (). This strongly suggests that the product is indeed LIMR, and it was thus purified for further analysis.
Figure 1. Expression and purification of LIMR. (A) S2 cells stably transfected with pMT/BiP/V5-His/LIMR express a ca. 42 kDa protein 1–4 days after induction (lanes labelled 1–4), which is not present in non-induced cells (lane labelled 0), as detected by Western blotting using α-V5-HRP. (B) LIMR purification on Ni Sepharose resin, as detected by Western blotting using α-V5-HRP: FT, flow-through; W, wash; E1–E6, elution fractions 1–6. (C) LIMR purification on α-V5-agarose resin results in a protein of high relative purity, as visualized by silver staining. The band in E2 was unambiguously identified as full-length human LIMR by MALDI-MS peptide mass fingerprinting. M, marker lane.
Purification of LIMR
Various detergents were tested for their ability to solubilize LIMR from S2 membranes: LDAO, Triton X-100, CHAPS and DDM. The use of the mild, non-ionic detergent DDM (Seddon et al. Citation2004) resulted in the largest fraction of LIMR present in the solubilized supernatant: approximately 60% as estimated by Western blotting. DDM is used therefore in all further experiments, at higher concentrations (1% [w/v]) to solubilize LIMR for purification and at lower concentrations (0.05–0.1% or 1–2 mM, well above the CMC of 0.18 mM (le Maire et al. Citation2000)) in all buffers for further functional and structural studies.
Solubilized LIMR was purified on Ni Sepharose, utilizing the interactions of the 6His-tag with chelated nickel ions. Almost all LIMR binds to the resin (), resulting in very little loss of protein. However, due to the low initial concentration of LIMR, and the non-specific binding of other proteins to the resin, the final eluate is an enriched but not pure receptor preparation. Therefore, as a second step, LIMR was purified on α-V5-agarose (), from which a protein preparation of high relative purity was eluted, as visualized by silver stained gels.
The protein construct of LIMR including its two tags has a calculated molecular mass of 59 kDa, yet shows an apparent molecular mass of ca. 42 kDa on SDS-PAGE. Although it is well known that membrane proteins often run anomalously on SDS-PAGE (Rath et al. Citation2009), it was important to unambiguously identify the purified product as full-length LIMR. Therefore, the silver stained band observed after α-V5-agarose purification () was analyzed by MALDI-MS peptide mass fingerprinting. Due to the high hydrophobicity of large parts of the protein, only 14% of the sequence of LIMR was detected by PMF. Nonetheless, human LIMR could be unambiguously identified as the only significant match for the peptide masses detected, and peptides corresponding to both the N- and C-termini of the intact receptor were found, showing that the protein was full-length and not truncated, despite its low apparent molecular mass (see ).
Figure 2. Peptide mass fingerprinting of LIMR. The silver stained band of purified protein (see ) was analyzed by MALDI/TOF-MS peptide mass fingerprinting. The sequence of LIMR (including tags) is shown, with the peptide fragments matched using the database search shown in bold and underlined. An additional peptide, which corresponds to the linker region between LIMR and the tags and is encoded by vector sequence, was matched manually and is shown underlined. Residues that constitute the nine putative transmembrane segments of LIMR are shown in bold italics. As is often the case, none of the transmembrane peptides were detected due to the hydrophobicity of these segments. The identified fragments, including peptide matches close to both the N- and C-termini, imply LIMR is present as the full-length receptor.

Oligomeric state of LIMR
A band corresponding in size to the dimeric form of LIMR was often observed on Western blots (), even when gels were run in the presence of SDS and β-mercaptoethanol. Further indications that LIMR might form dimers or higher order oligomers came from crosslinking and co-immunoprecipitation experiments. Therefore, the oligomeric state of the receptor was further analyzed by gel filtration. A Ni Sepharose-purified, concentrated solution of LIMR in mild buffer containing DDM was applied to a column of Superose 6 resin. Eluted fractions were analyzed for the presence of LIMR by SDS-PAGE and subsequent Western blotting (). Quantification of the relative amounts of LIMR in each fraction by densitometry resulted in an elution profile (), which was compared to a calibration curve to estimate the molecular mass and thereby aggregation number of the eluted LIMR complexes.
Figure 3. LIMR forms dimers and larger oligomers in DDM-solubilized form. (A) DDM-solubilized, semi-purified LIMR was separated by gel filtration on a Superose 6 column. Eluted fractions (60 fractions of 0.5 ml each) were collected and analyzed for the presence of LIMR by SDS-PAGE and subsequent Western blotting using α-V5-HRP. All LIMR was present in fractions 11–27 (lanes F11–27). (B) The relative amount of the receptor in each fraction was quantified using AIDA 2D densitometry software, resulting in the elution profile shown in the graph (light grey curve, left y-axis). A calibration curve was constructed based on the elution volumes of marker proteins (grey circles), and shows the linear relationship between the log of the molecular mass (right y-axis) and the apparent partition coefficient K av. Four distinct peaks are present in the LIMR elution profile: The major peak elutes last, in F23 and 24 or at a K av of 0.55, and presumably corresponds to dimeric LIMR (144 kDa or 2.4 times the LIMR protomer). Two additional peaks are observed that elute earlier, i.e., have a larger molecular mass, and correspond to LIMR oligomers. The small peak eluting first around F12 consists of very large aggregates (aggregation number > 100).
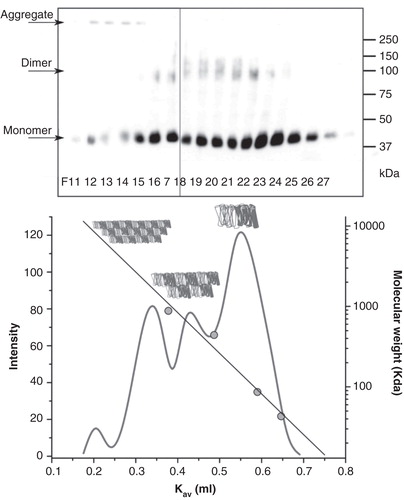
The major part of LIMR elutes around fractions 23 and 24, corresponding to a partition coefficient K av of approximately 0.55. This species has a molecular mass of ca. 144 kDa, approximately 2.4 times the molecular mass of the LIMR protomer, and is thus most likely the receptor dimer, given that the protein will also bind a significant amount of detergent and the calibration was carried out using water-soluble proteins. Two separate peaks of higher molecular mass elute around F19/20 and F16/17, corresponding to LIMR oligomers. Finally, a very small amount of large aggregates elutes around F12.
SPR studies of LIMR-ligand interactions
One of the main questions concerning LIMR is its ligand-binding behaviour: First identified as a specific receptor for Lcn1, LIMR was later reported to bind pVEG, UG and BLG as well, but neither the exact mechanism nor the biological significance of these interactions is clear. Therefore, the binding of all four proposed ligands to the receptor was studied qualitatively and quantitatively by surface plasmon resonance. To this end, an antibody directed towards the V5-tag was covalently immobilized on the surface of an SPR-chip, and LIMR was subsequently captured from a purified, solubilized receptor preparation (Supplementary Figure S2, available online). Ligand binding was measured in single cycle kinetics (SCK) experiments (Karlsson et al. Citation2006), a method that uses a single surface, with immobilized antibody and captured receptor, to flow over five increasing concentrations of analyte, followed by a long dissociation phase. Kinetic parameters k on and k off were then obtained from fits to the association and dissociation phases, respectively, of the corrected binding curves.
SCK binding of Lcn1 to LIMR was studied using a range of analyte concentrations, from 0.13, 0.25, 0.5, 1.0, 2.0 μM to 3.1, 6.3, 13, 25, 50 μM. Response levels ranged from ca. 3 RU for the lowest concentration to ca. 60 RU for the highest. Binding to the reference flow cell was absent, bar the sharp changes at the beginning and end of injection; this reference signal was used for correction. During buffer injections, a slow loss of receptor from the surface was observed due to dissociation of the antibody-V5-tag binding, of approximately 1 RU/min, which was also corrected for. shows a representative binding curve with fit of SCK binding of Lcn1 to LIMR. K d values of 0.17–3.9 μM were obtained from these experiments (), although these values may be a little too high due to mass transport limitations (see Discussion). Since the molecular mass or surface density of the receptor is not a parameter in the SCK model (Karlsson et al. Citation2006), the oligomerization of LIMR does not influence these results.
Figure 4. LIMR is a specific receptor for Lcn1, and does not exhibit high affinity for pVEG, UG or BLG. (A) SPR response curve (grey) showing the binding of Lcn1 to LIMR in a single cycle kinetics experiment, injecting 5 times 60-second (30 μl) pulses of a 2-fold concentration range of Lcn1 (1.3, 2.5, 5.0, 10, 20 μM), followed by a long dissociation phase. The response curve was fitted (black) to derive kinetic parameters from the data (). (B) SPR response curves showing the binding of UG (black, up to 0.5 μM), pVEG (grey, up to 13 μM) and BLG (black dashed, up to 50 μM) to LIMR in single cycle kinetics experiments. None of these proposed ligands interact with the receptor. The presence and activity of LIMR on the chip surface was confirmed after each experiment by injection of Lcn1, which bound as expected in all cases. Doubly corrected data are shown in both panels, after subtraction of the signal from a reference flow cell, and of the signal from 5 times 60-second pulses of buffer injection over the receptor flow cell.
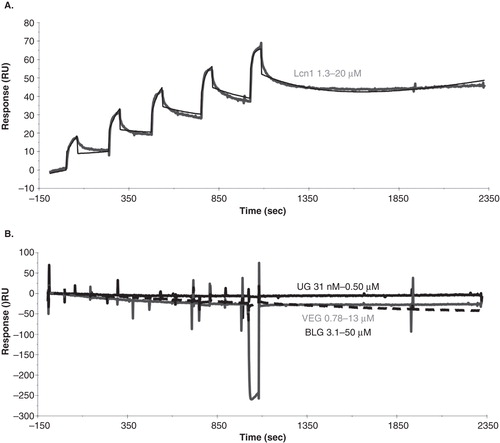
Table I. Experimental variables and kinetic parameters for the binding of Lcn1 to LIMR from four independent SCK experiments.
depicts representative binding curves of SCK binding of pVEG, UG and BLG to LIMR. None of these proposed ligands interacted with LIMR under the conditions used, up to analyte concentrations of 50 μM. After each SCK measurement, Lcn1 was injected and shown to bind, to prove the presence and activity of the receptor on the chip surface. These data can be interpreted in two ways: Either pVEG, UG and BLG are not ligands for LIMR, or they bind to a site on the receptor distinct from the Lcn1 binding site, in which case the former site could be unexposed, inaccessible or inactive.
Discussion
In this study, we developed a system for the expression, solubilization and purification of LIMR, the first specific lipocalin receptor to be identified (Wojnar et al. Citation2001) and the prototype of a new family of membrane proteins (Sivaprasadarao and Findlay Citation1988a, Citation1988b, Wojnar et al. Citation2003, Kawaguchi et al. Citation2007). The receptor is shown to form oligomers in DDM-solubilized state, probably mostly dimers, but very little aggregates. We subsequently addressed the question whether LIMR is a high-affinity receptor solely for Lcn1, as was first thought (Wojnar et al. Citation2001, Citation2003), or rather a more promiscuous receptor, binding also pVEG (Burke Citation2004), UG (Zhang et al. Citation2006) and BLG (Fluckinger et al. Citation2008). The SPR single cycle kinetics data strongly suggest that the former hypothesis is correct: LIMR only interacts with Lcn1.
The determination of the size of membrane proteins and membrane protein complexes is complicated by the fact that these proteins often behave differently from soluble size markers, for example with respect to detergent binding (Rath et al. Citation2009). In SDS-PAGE, gel shifts of approximately 50% have been reported (Rath et al. Citation2009), in both directions (Wegener and Jones Citation1984, Neumann et al. Citation1998). A negative gel shift of −29% is observed for LIMR, which could indicate relatively low levels of SDS loading, possibly caused by the replacement of protein-detergent interactions by intramolecular protein-protein contacts. The mass spectrometry data rule out any proteolytically derived loss of mass.
In gel filtration, the partition coefficient K av depends on the hydrodynamic volume of the particle, described by its Stokes radius or viscosity radius (Irvine Citation2001). Membrane proteins are expected to bind a much larger number of detergent molecules than water-soluble proteins, which would lead to an overestimation of their hydrodynamic radii (le Maire et al. Citation1986). For this reason, the results presented here for LIMR are not definitive, but the most reasonable interpretation is that the receptor forms dimers, especially since this species is also observed by other techniques, e.g., Western blotting.
The SPR studies show that LIMR is a high-affinity receptor for Lcn1, the lipocalin that was used as bait to identify the receptor. This finding disagrees with literature reports describing interactions with Lcn1's porcine homolog pVEG (Burke Citation2004), the bovine lipocalin BLG (Fluckinger et al. Citation2008) and the secretoglobin UG (Zhang et al. Citation2006). The literature data suggesting interactions between LIMR and pVEG are all based on SPR studies, using HEK293 membranes containing native LIMR, or short synthetic peptides mimicking extracellular domains of the receptor (Burke Citation2004). The published data exhibit high levels of non-specific binding as compared to the specific response, and are therefore difficult to interpret.
Interactions between LIMR and the bovine lipocalin BLG have been studied using a short N-terminal LIMR fragment expressed in E. coli and purified without the use of detergents (Fluckinger et al. Citation2008). This fragment contains putative eukaryotic transmembrane segments, which might be prone to aggregation or non-specific hydrophobic contacts. In addition, FITC-labelled BLG is internalized by NT2 cells, and by Sf9 cells recombinantly expressing LIMR, but to a lesser extent by LIMR-antisense transfected NT2 cells or untransfected Sf9 cells (Fluckinger et al. Citation2008). These fluorescence microscopy data tend to support specific interactions between LIMR and BLG, but again background signals were significant.
The complete lack of binding in our SPR studies, despite injecting BLG up to a concentration of 50 μM, strongly suggests that the bovine lipocalin is not a high-affinity ligand for LIMR. Lcn1 and BLG are similar in sequence (Redl Citation2000) and structure (Papiz et al. Citation1986, Breustedt et al. Citation2005) (27% identity on the protein sequence level, and a root mean square deviation for the C-α positions of the β-barrel of 1.7 Å (Breustedt et al. Citation2005)), and thus could at least partially share a binding site on the relatively small extracellular domain of LIMR (Wojnar et al. Citation2003), especially since both have been suggested to bind to the N-terminus (Wojnar et al. Citation2001, Fluckinger et al. Citation2008). The binding site for Lcn1 is well exposed, correctly folded and active in the SPR experiments, as verified by the binding curves obtained when injecting Lcn1. Therefore, the lack of response observed when injecting BLG is a significant indication that LIMR is not a classical high-affinity receptor for this bovine lipocalin. Due to the high degree of similarity between Lcn1 and BLG, and between LIMR and its bovine orthologs (Wojnar et al. Citation2003), there might be a low degree of coincidental affinity of LIMR for BLG, which could explain the fluorescence microscopy data in the literature but which was not detected in the SPR studies presented here. It is unlikely that this binding behaviour is physiologically significant, although it may contribute to BLG's role as a food-borne allergen (Fluckinger et al. Citation2008).
Finally, radiolabelled UG was shown to bind to LIMR-expressing COS-1, HTB-30, HTB-81 and HTB-174 cells in a specific manner, with high affinity (K d = 18 nM for binding to HTB-81 cells) (Zhang et al. Citation2006). We obtained no response in the SPR studies, despite injecting UG at a concentration 50 times higher than the reported K d. UG is unrelated to the lipocalins and completely different in structure (Mukherjee et al. Citation2008), so it might bind to a different site on the receptor, which could be unexposed, inaccessible or misfolded. Alternatively, UG is simply not a ligand for LIMR. Interestingly, binding of UG to a different receptor, a GPCR, was also reported (Ray et al. Citation2006).
When comparing the various sets of experimental data, it should be borne in mind that the apo- or holo-state of small molecule-binding proteins can be decisive for receptor interactions, as is the case for RBP and its receptor, and for 24p3. Especially for the more promiscuous lipocalins, such as Lcn1 and BLG, which bind many different ligands with moderate affinities, knowledge of the ligand-binding state is not always available. In this work, the lipocalins are at least partly in the holo-state: Lcn1 and pVEG have acquired ligands from the bacterial cells or media, and the commercially acquired BLG was sourced from bovine milk, and contains endogenous ligands. Pre-incubation with a 5 times molar excess of retinol to bring the lipocalins fully in the holo-state made no difference to the binding behaviour in this study, although both Lcn1 and BLG bind this ligand. However, the influence of the apo- or holo-state of lipocalins on their receptor binding behaviour is a subject that merits further investigation, and that could explain some of the conflicting experimental data, since different ligands can induce different binding characteristics (Sivaprasadarao and Findlay Citation1988a, Citation1988b, Redondo et al. Citation2008).
Besides the state of the lipocalins, the activation state of the receptor can also influence the binding behaviour. Receptors may need interactions with membrane lipids, the cytoskeleton, or other proteins in order to be able to retain native binding characteristics. Literature studies have used peptide fragments, recombinantly expressed proteins, and native membranes to study the behaviour of LIMR. In this study, recombinantly expressed and purified LIMR is active and fully able to bind Lcn1. However, binding of the other presumed ligand via a different mechanism for which the receptor needs a different environment cannot be completely excluded.
The interaction between LIMR and its ligand Lcn1 was measured quantitatively as well as qualitatively, and K d values of 0.17–3.9 μM were obtained. The accuracy of the kinetics and affinity data for the LIMR-Lcn1 interaction mainly depends on the influence of mass transport effects on these measurements. Such effects occur if the diffusion between the bulk flow and the surface is slow as compared to the association and dissociation events taking place at this surface (Rich and Myszka Citation2000). The observation that an increase in flow rate from 30–60 μl/min results in a pronounced increase in apparent k on and decrease in apparent K d () indicates that the binding is indeed mass transport limited. The actual K d is thus likely to be closer to this lower value of 0.17 μM than to the higher values measured. However, the high variability in the quantitative data is reflected in the large error bar on this value for K d.
The ligand-binding behaviour of LIMR will be intimately linked to its structure and biological function. The literature data showing internalization of many different ligands with low specificity (Wojnar et al. Citation2003, Zhang et al. Citation2006, Fluckinger et al. Citation2008), indicate a function similar to that of megalin (Christensen and Birn Citation2002), in the uptake of a broad range of proteins with bound ligands. However, the putative topology proposed for LIMR is very different from that of a typical endocytic receptor: whereas the latter often has a single transmembrane segment and a large extracellular domain, LIMR has nine putative transmembrane segments and a relatively small extracellular domain (Wojnar et al. Citation2001, Citation2003). It is perhaps noteworthy that a similar topography has been proposed for the RBP receptor STRA6 which is also the transporter for the RBP ligand retinol. The highly specific interaction with Lcn1 observed in this study, hints at a more distinctive function for LIMR, complementing that of Lcn1. Hypothesizing on the receptor's function is complicated by the fact that the exact role of Lcn1 is still unclear. One of the main roles of the lipocalin is presumably scavenging of hydrophobic, potentially harmful molecules (Glasgow et al. Citation1995, Lechner et al. Citation2001, Gasymov et al. Citation2005), and antimicrobial action (Fluckinger et al. Citation2004). LIMR could thus be involved in the uptake of molecules bound to Lcn1, in the detoxification of harmful compounds, and in the sequestering of useful ligands, such as vitamins (Glasgow et al. Citation2002) or iron-loaded siderophores (Goetz et al. Citation2002, Fluckinger et al. Citation2004, Devireddy et al. Citation2005).
Conclusions
LIMR, the first specific lipocalin receptor to be identified and the prototype of a new family of membrane proteins, was expressed in S2 cells and purified by affinity chromatography. The receptor forms dimers or larger oligomers when solubilized in DDM. The full-length, functional receptor, captured onto a surface plasmon resonance chip via an α-V5 antibody, interacts specifically with Lcn1, binding the lipocalin with low micromolar to high nanomolar affinity. No interactions with any of the other putative ligands could be detected, raising doubts about the physiological relevance of the reported binding of pVEG, BLG and UG to the receptor.
Supplementary Figures S1 and S2
Download MS Word (161.5 KB)Acknowledgements
This work was funded by a Marie Curie Early Stage Training Fellowship from the European Community's Sixth Framework Program under contract MEST-CT-2004-007931-BIOMEM. We are very grateful to Berhard Redl (Department of Molecular Biology, University of Innsbruck, Austria) for providing the LIMR coding sequence.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Akerstrom B, Borregaard N, Flower DR, Salier JP. 2006. Lipocalins. Landes Bioscience/Eurekah.com.
- Akerstrom B, Flower DR, Salier JP. 2000. Lipocalins: Unity in diversity. Biochim Biophys Acta 1482:1–8.
- Breton J, Gage MC, Hay AW, Keen JN, Wild CP, Donnellan C, et al. 2008. Proteomic screening of a cell line model of esophageal carcinogenesis identifies cathepsin D and aldo-keto reductase 1C2 and 1B10 dysregulation in Barrett's esophagus and esophageal adenocarcinoma. J Proteome Res 7:1953–1962.
- Breustedt DA, Korndorfer IP, Redl B, Skerra A. 2005. The 1.8-angstrom crystal structure of human tear lipocalin reveals an extended branched cavity with capacity for multiple ligands. J Biol Chem 280:484–493.
- Briand L, Trotier D, Pernollet JC. 2004. Aphrodisin, an aphrodisiac lipocalin secreted in hamster vaginal secretions. Peptides 25:1545–1552.
- Burke B. 2004. Characterisation of the lipocalin-1 receptor system. School of Biochemistry and Microbiology University of Leeds. Leeds, UK.
- Burke BJ, Redondo C, Redl B, Findlay JBC. 2005. Lipocalin receptors: Into the spotlight. In: Åkerström B, Borregaard N, Flower DR, Jean-Philippe Salier J-P, editors. Lipocalins. Austin, TX: Landes Bioscience.
- Christensen EI, Birn H. 2002. Megalin and cubilin: Multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3:256–266.
- Clagett-Dame M, DeLuca HF. 2002. The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr 22:347–381.
- Devireddy LR, Gazin C, Zhu XC, Green MR. 2005. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123:1293–1305.
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432:917–921.
- Flower DR. 2000a. Beyond the superfamily: The lipocalin receptors. Biochim Biophys Acta 1482:327–336.
- Flower DR. 2000b. Experimentally determined lipocalin structures. Biochim Biophys Acta 1482:46–56.
- Flower DR, North ACT, Sansom CE. 2000. The lipocalin protein family: Structural and sequence overview. Biochim Biophys Acta 1482:9–24.
- Fluckinger M, Haas H, Merschak P, Glasgow BJ, Redl B. 2004. Human tear lipocalin exhibits antimicrobial activity by scavenging microbial siderophores. Antimicrob Agents Ch 48:3367–3372.
- Fluckinger M, Merschak P, Hermann M, Haertlé T, Redl B. 2008. Lipocalin-interacting-membrane-receptor (LIMR) mediates cellular internalization of beta-lactoglobulin. Biochim Biophys Acta 1778:342–347.
- Fullard RJ, Tucker DL. 1991. Changes in human tear protein levels with progressively increasing stimulus. Invest Ophth Vis Sci 32:2290–2301.
- Gasymov OK, Abduragimov AR, Prasher P, Yusifov TN, Glasgow BJ. 2005. Tear lipocalin: Evidence for a scavenging function to remove lipids from the human corneal surface. Invest Ophth Vis Sci 46:3589–3596.
- Glasgow BJ, Abduragimov AR, Farahbakhsh ZT, Faull KF, Hubbell WL. 1995. Tear lipocalins bind a broad array of lipid ligands. Curr Eye Res 14:363–372.
- Glasgow BJ, Abduragimov AR, Gassymov OK, Yusifov TN, Ruth EC, Faull KF. 2002. Vitamin E associated with the lipocalin fraction of human tears. Adv Exp Med Biol 506:567–572.
- Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. 2002. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10:1033–1043.
- Gouveia SM, Tiffany JM. 2005. Human tear viscosity: An interactive role for proteins and lipids. Biochim Biophys Acta 1753:155–163.
- Irvine GB. 2001. Determination of molecular size by size-exclusion chromatography (gel filtration). Curr Protoc Cell Biol; 5.5.1–5.5.16.
- Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. 2006. Analyzing a kinetic titration series using affinity biosensors. Anal Biochem 349:136–147.
- Kawaguchi R, Yu JM, Honda J, Hu J, Whitelegge J, Ping PP, et al. 2007. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315:820–825.
- Kontopidis G, Holt C, Sawyer L. 2002. The ligand-binding site of bovine beta-lactoglobulin: Evidence for a function? J Mol Biol 318:1043–1055.
- Kontopidis G, Holt C, Sawyer L. 2004. Beta-lactoglobulin: Binding properties, structure, and function. J Dairy Sci 87:785–796.
- le Maire M, Aggerbeck LP, Monteilhet C, Andersen JP, Moller JV. 1986. The use of high-performance liquid chromatography for the determination of size and molecular weight of proteins: A caution and a list of membrane proteins suitable as standards. Anal Biochem 154:525–535.
- le Maire M, Champeil P, Moller JV. 2000. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim Biophys Acta 1508:86–111.
- Lechner M, Wojnar P, Redl B. 2001. Human tear lipocalin acts as an oxidative-stress-induced scavenger of potentially harmful lipid peroxidation products in a cell culture system. Biochem J 356:129–135.
- Millar TJ, Mudgil P, Butovich IA, Palaniappan CK. 2009. Adsorption of human tear lipocalin to human meibomian lipid films. Invest Ophth Vis Sci 50:140–151.
- Morize I, Surcouf E, Vaney MC, Epelboin Y, Buehner M, Fridlansky F, et al. 1987. Refinement of the C2221 crystal form of oxidized uteroglobin at 1.34 A resolution. J Mol Biol 194:725–739.
- Mukherjee AB, Zhang Z, Chilton BS. 2008. Uteroglobin: A steroid-inducible immunomodulatory protein that founded the secretoglobin superfamily. Endocr Rev 29:131–131.
- Neumann S, Matthey U, Kaim G, Dimroth P. 1998. Purification and properties of the F1F0 ATPase of Ilyobacter tartaricus, a sodium ion pump. J Bacteriol 180:3312–3316.
- O’Connell KL, Stults JT. 1997. Identification of mouse liver proteins on two-dimensional electrophoresis gels by matrix-assisted laser desorption/ionization mass spectrometry of in situ enzymatic digests. Electrophoresis 18:349–359.
- Papiz MZ, Sawyer L, Eliopoulos EE, North ACT, Findlay JBC, Sivaprasadarao R, et al. 1986. The structure of beta-lactoglobulin and its similarity to plasma retinol-binding protein. Nature 324:383–385.
- Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, et al. 1999. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J 18:4633–4644.
- Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. PNAS 106:1760–1765.
- Ray R, Zhang Z, Lee YC, Gao JL, Mukherjee AB. 2006. Uteroglobin suppresses allergen-induced TH2 differentiation by down-regulating the expression of serum amyloid A and SOCS-3 genes. FEBS Lett 580:6022–6026.
- Redl B. 2000. Human tear lipocalin. Biochim Biophys Acta 1482:241–248.
- Redondo C, Vouropoulou M, Evans J, Findlay JBC. 2008. Identification of the retinol-binding protein (RBP) interaction site and functional state of RBPs for the membrane receptor. FASEB J 22:1043–1054.
- Rich RL, Myszka DG. 2000. Advances in surface plasmon resonance biosensor analysis. Curr Opin Biotech 11:54–61.
- Seddon AM, Curnow P, Booth PJ. 2004. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim Biophys Acta 1666:105–117.
- Sivaprasadarao A, Findlay JBC. 1988a. The interaction of retinol-binding protein with its plasma-membrane receptor. Biochem J 255:561–569.
- Sivaprasadarao A, Findlay JBC. 1988b. The mechanism of uptake of retinol by plasma-membrane vesicles. Biochem J 255:571–579.
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. 1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150:76.
- Wegener AD, Jones LR. 1984. Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. Evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J Biol Chem 259:1834–1841.
- Wojnar P, Lechnar M, Merschak P, Redl B. 2001. Molecular cloning of a novel lipocalin-1 interacting human cell membrane receptor using phage display. J Biol Chem 276:20206–20212.
- Wojnar P, Lechner M, Redl B. 2003. Antisense down-regulation of lipocalin-interacting membrane receptor expression inhibits cellular internalization of lipocalin-1 in human NT2 cells. J Biol Chem 278:16209–16215.
- Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. 2005. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436:356–362.
- Zhang ZJ, Kim SJ, Chowdhury B, Wang JY, Lee YC, Tsai PC, et al. 2006. Interaction of uteroglobin with lipocalin-1 receptor suppresses cancer cell motility and invasion. Gene 369:66–71.