Abstract
Epidemiological studies indicate that patients suffering from atherosclerosis are predisposed to develop osteoporosis. Accordingly, atherogenic determinants such as oxidized low density lipoprotein (OxLDL) particles have been shown to alter bone cell functions. In this work, we investigated the cytotoxicity of lysophosphatidylcholine (lysoPC), a major phospholipid component generated upon LDL oxidation, on bone-forming MG-63 osteoblast-like cells. Cell viability was reduced by lysoPC in a concentration-dependent manner with a LC50 of 18.7 ± 0.7 μM. LysoPC-induced cell death was attributed to induction of both apoptosis and necrosis. Since impairment of intracellular calcium homeostasis is often involved in mechanism of cell death, we determined the involvement of calcium in lysoPC-induced cytotoxicity. LysoPC promoted a rapid and transient increase in intracellular calcium attributed to mobilization from calcium stores, followed by a sustained influx. Intracellular calcium mobilization was associated to phospholipase C (PLC)-dependent mobilization of calcium from the endoplasmic reticulum since inhibition of PLC or calcium depletion of reticulum endoplasmic with thapsigargin prevented the calcium mobilization. The calcium influx induced by lysoPC was abolished by inhibition of transient receptor potential vanilloid (TRPV) channels with ruthenium red whereas gadolinium, which inhibits canonical TRP (TRPC) channels, was without effect. Accordingly, expression of TRPV2 and TRPV4 were shown in MG-63 cells. The addition of TRPV2 inhibitor Tranilast in the incubation medium prevent the calcium influx triggered by lysoPC and reduced lysoPC-induced cytotoxicity whereas TRPV4 inhibitor RN 1734 was without effect, which confirms the involvement of TRPV2 activation in lysoPC-induced cell death.
Introduction
Epidemiologic studies have reported a correlation between atherosclerosis and the development of osteoporosis (Uyama et al. Citation1997, Barengolts et al. Citation1998). Indeed, it was shown that patients with lower bone density and osteoporosis present higher lipid levels (Uyama et al. Citation1997, Barengolts et al. Citation1998). It is well established that cardiovascular diseases are directly related to plasma concentrations of LDL cholesterol (Castelli et al. Citation1977), which suggests that both pathologies may shear one or several contributory factors. Accordingly, atherosclerosis-susceptible C57BL/6 mice maintained under an atherogenic high-fat diet show reduced bone formation and low bone mass (Parhami et al. Citation2001).
Osteoporosis results from the loss of equilibrium between bone cell functions, such as increased osteoclast-mediated bone resorption over bone formation by osteoblasts, leading to reduced bone mass, lower mineral density, and thereby higher bone fragility and susceptibility to fractures. Although the importance of cholesterol in the development of osteoporosis has been highlighted, it remains largely unknown how lipids can affect bone metabolism. Given that it is well established that LDL particles become pro-atherogenic after undergoing oxidative modifications (Steinberg Citation1997), oxidized LDL (OxLDL) would be expected to be also responsible for osteoporosis. Indeed, studies have indicated that OxLDL have deleterious effects on the differentiation of cells towards an osteoblastic phenotype (Parhami et al. Citation1997, Mody et al. Citation2001). OxLDL and oxidative products inhibit osteogenic differentiation of mesenchymal stem cells and preosteoblast in favour of an adipogenic phenotype. Also, studies showed that OxLDL caused cell death of human osteoblasts (Klein et al. Citation2003, Citation2006). The possible implication of OxLDL in the development of osteoporosis was further highlighted by our studies which demonstrated that osteoblastic cells express receptors for OxLDL (Brodeur et al. Citation2008a) and that OxLDL particles promote cell death of osteoblasts (Brodeur et al. Citation2008b, Hamel et al. Citation2008).
Upon oxidative modifications of LDL, oxysterols are generated such as 7-ketocholesterol and 7β-hydroxycholesterol which are found in atherosclerotic lesions (Brown and Jessup Citation1999). Moreover, phosphatidylcholine is converted to lysophosphatidylcholine (lysoPC) by phospholipase A2 activity present in LDL (Parthasarathy et al. Citation2010). Thereby, lysoPC is a major phospholipid component of OxLDL and was identified as a critical factor in the atherogenic activity of OxLDL (Schmitz and Ruebsaamen Citation2010). LysoPC has been associated with atherosclerosis, as well as acute and chronic inflammation by altering various functions in a number of cell types including endothelial cells, smooth muscle cells, monocytes, macrophages, and lymphocyte T-cells (Fuchs and Schiller Citation2009). Among others, lysoPC triggers apoptosis of vascular smooth muscle cells (Hsieh et al. Citation2000, Hsu et al. Citation2011) and endothelial cells (Takahashi et al. Citation2002, Matsubara and Hasegawa Citation2005, Zhou et al. Citation2006) by altering intracellular calcium signaling (Zhou et al. Citation2006, Song et al. Citation2010, Hsu et al. Citation2011). Mechanistic evidence was given by studies showing that activation of transient receptor potential channels was associated to oxidative stress-induced cell death (Miller Citation2006) and to OxLDL-induced apoptosis of vascular smooth muscle cells (Ingueneau et al. Citation2009).
The transient receptor potential (TRP) channels contain six putative transmembrane domains, which are thought to assemble as homo- or hetero-tetramers to form channels (for a review, Pedersen et al. Citation2005). All TRPs are cation channels, although the permeability for different mono- and divalent cations varies greatly between isoforms. They are activated by a wide range of stimuli including intra- and extracellular messengers, chemical, mechanical, and osmotic stress, and some by the filling state of intracellular calcium stores. Based on amino acid homologies, the mammalian TRP channel superfamily has been divided into seven families: TRPC, TRPM, TRPV, TRPML, TRPP, TRPA and TRPN. The TRPC (‘Canonical') and TRPM (‘Melastatin') subfamilies consist of seven and eight different channels, respectively (i.e., TRPC1–TRPC7 and TRPM1–TRPM8). The TRPV (‘Vanilloid') subfamily presently comprises six members (TRPV1–TRPV6). The TRPML (‘Mucolipin') family comprises three members, and the TRPP (‘Polycystin') family three channel-like and five non-channel members, respectively. The most recently proposed subfamily, TRPA (‘Ankyrin'), has only one mammalian member, TRPA1, and finally, the TRPN (no mechanoreceptor potential C, or NOMPC) has so far only been detected in Caenorhabditis elegans, Drosophila and zebra fish. There has been increasing interest in first three families because of their involvement in several human diseases while last four families are yet insufficiently characterized. We have reported the expression of numerous TRP channels in osteoblastic cells (Abed and Moreau Citation2007, Labelle et al. Citation2007, Abed et al. Citation2009).
In the current study, we investigated the cytotoxicity of lysoPC as a major phospholipid component generated upon LDL oxidation, on human osteoblast-like MG-63 cells and determined the involvement of calcium in lysoPC-induced cytotoxicity.
Materials and methods
Cell culture
Human osteoblast-like MG-63 cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and were grown in a 1:1 mixture of phenol-free DMEM/Ham's F12 medium (DMEM/F12; Sigma, Oakville, Ontario, Canada) supplemented with 10% fetal bovine serum (FBS; Cansera, Etobicoke, Ontario, Canada), L-glutamine (Invitrogen, Burlington, Ontario, Canada) and penicillin/streptomycin (Invitrogen). Cells were cultured in 5% CO2 at 37°C and were harvested weekly with Trypsin-EDTA solution (Invitrogen).
Cell viability assays
For cell viability experiments, cells were seeded in 96-well plates (Sarstedt, Montréal, Québec, Canada). After five days of culture in supplemented media, cells were incubated in culture medium without serum in the absence or the presence of increasing concentrations of lysoPC (Sigma) or cannabidiol (Tocris Bioscience, UK) without or with gadolinium (Gd3+), ruthenium red (Sigma), RN 1734 (Sigma) and Tranilast (Sigma) for 48 h. Experiments were also performed with cells treated for 48 h with GSK1016790A (Sigma). For experiments with low levels of calcium in medium, cells were incubated in DME/F12 (culture medium without calcium and magnesium, Sigma) supplemented with 0.25 mM calcium and 0.8 mM magnesium. Cell viability was determined by microtiter tetrazolium reduction assays. Briefly, 1 h before the end of treatment the medium was replaced with DMEM/F12 containing 0.5 mg/ml of 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrasodium bromide (MTT) (Sigma). At the end of the incubation, media was aspired and formazan crystals generated by the cellular reduction activity were dissolved in dimetylsulfoxide (DMSO). Absorbance was measured at 575 nm and data are expressed as the ratio of absorbance of treated cells versus control cells. In parallel experiments, cells exposed to lysoPC were dyed with crystal violet, then solubilized in 1% SDS and quantified with a spectrophotometer (575 nm), or harvested by trypsinization to perform cell counts.
Cell death assays
MG-63 cells seeded in 12-well plates were incubated in culture medium alone or with lysoPC, as well as in the presence of ruthenium red or Tranilast. After 24 h, floating cells were discarded and adherent cells were collected by trypsinization and centrifugation at 250 g for 5 min. Cells were washed with PBS and then incubated with 2 μg of propidium iodide (PI) and 10 μl of FITC-annexin V per milliter (Roche Diagnostics, Mannheim, Germany) for 15 min at 4°C. At the end of incubation, 400 μl of cold PBS was added and cellular FITC-annexin V and PI fluorescences were detected by using a fluorescence-activated cell sorter (FACScan, Becton Dickinson, Mississauga, Canada) and data were analyzed with Cell Quest software (Becton Dickinson). Cells positive for FITC-annexin V were considered apoptotic whereas cells positive for PI and negative for FITC-annexin V were considered necrotic.
Reverse-transcription polymerase chain reaction
Total RNA from cells was extracted using TriZol (Invitrogen) according to the manufacturer's instructions. Reverse transcription (RT) reactions were carried out with Omniscript RT kit (Qiagen, Mississauga, Ontario, Canada) using hexamers. The PCR amplifications were conducted with Taq PCR core kit (Qiagen) using specific primer sets for human TRPV1-6 (Abed et al. Citation2009). Each primer was designed in distinct exons to ensure specific transcript amplifications. Briefly, amplifications were carried out for 40 cycles according to incubation of 1 min at 94°C, 30 sec at 58°C and 1 min at 72°C. Amplification products were resolved in 2% agarose gel with ethidium bromide revelation.
Measurements of intracellular calcium
MG-63 cells were cultured in 8-wells Labtek (Nalge Nunc, Napperville, IL, USA) for five days in supplemented media. Cells were then transferred to HEPES-buffered saline solution (HBSS (mM): 121 NaCl, 5.4 KCl, 25 HEPES, 1.8 CaCl2 and 6.0 NaHCO3 at pH 7.3) and loaded with 2 μM Fluo-3 AM with an equivalent volume of 20% Pluronic F127 (Invitrogen) for 45 min at room temperature in the dark. Thereafter cells were washed with corresponding HBSS and the loaded dye was allowed to de-esterify for 45 min at room temperature in the dark. Following transfer to a calcium-free HBSS, treatments were made in an open chamber configuration at room temperature. The cells were examined with a laser scanning confocal (Bio-Rad) microscope (Nikon TE300) with an Apochromatic 40× N.A. 1.0 objective lens. Fluorescence was excited by an argon laser at 488 nm and emission was collected with a 515 nm filter. Data were analyzed with Laser Sharp 2.1T, Time Course 1.0 software for 7–8 fields per experiments (between 10 and 20 cells per field). All experiments were performed for at least four individual experiments.
Immunolocalization
MG-63 cells were cultured in eight-well Labtek (Nalge Nunc) dishes for five days in supplemented medium. Cells were fixed in 4% formaldehyde/phosphate-buffered saline (PBS) for 30 min at 4°C, permeabilized with 0.05% Tween/PBS for 15 min and blocked in 2% bovine serum albumin/PBS for 30 min. Cells were incubated overnight with primary antibodies (1:500 for anti-TRPV2 and anti-TRPV4 from Alomone, Jerusalem, Israel) or primary antibodies preincubated with 1 μg antigen followed by an incubation with secondary antibodies (1:2000 for anti rabbit IgG coupled to rhodamine from Chemicon, Temecula, CA) for 1 h. Cells were washed three times with PBS between each step. The cells were examined with a laser scanning confocal (Bio-Rad) microscope (Nikon TE300) with an Apochromatic 40× N.A. 1.0 objective lens. Fluorescence was excited by an argon laser at 555 nm and emission was collected with a 565 filter.
Statistical analysis
Data is expressed as mean ± SEM, and the significance of differences between groups has been determined using GrafPadPrism 5.0 Software by analysis of variance. Differences between groups were further evaluated by Student's t-test or two-way ANOVA with Bonferroni or Dunnett post tests. Differences were considered significant at p ≤ 0.05.
Results
Effect of LysoPC on the viability of MG-63
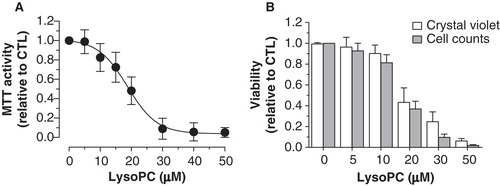
Our previous studies showed that OxLDL particles cause osteoblastic cell death (Hamel et al. Citation2008, Brodeur et al. Citation2008b). Since lysoPC is a major phospholipid component generated upon LDL oxidation (Parthasarathy et al. Citation2010), we determined its effect on the viability of osteoblastic MG-63 cells. We performed MTT assays on cells incubated for 48 h with increasing concentrations of lysoPC. As shown in , exposition of MG-63 cells to lysoPC resulted in a reduction of the MTT activity in a concentration-dependent manner. Curve analysis with the Boltzmann sigmoidal equation give a LC50 (concentration that cause 50% reduction of MTT activity) of 18.7 ± 0.7 μM with a significant reduction starting from 15 μM (Dunnett test, p < 0.05). To ascertain that this reduction of MTT activity corresponded to cell mortality, similar experiments were performed and remaining adherent cells were either stained with crystal violet or counted after trypsinization. In accordance with data from MTT assays, similar dose-dependent reductions of crystal violet staining and cell counts were obtained () for cells treated with increasing concentrations of lysoPC with LC50 values of 17.6 ± 1.4 and 16.5 ± 0.9 μM, respectively. These different experiences confirmed that lysoPC reduces MG-63 cell viability in a dose-dependent manner.
Figure 1. Cytotoxicity of lysoPC in MG-63 cells. (A) Dose-response curve of MTT activity (relative to control without lysoPC) following 48-h exposure to increasing concentrations of lysoPC in serum-free culture medium. Values are mean ± SEM of five independent experiments performed in triplicates. (B) Dose-response analysis of cell viability (relative to control without lysoPC) following 48-h exposure to increasing concentrations of lysoPC in serum-free culture medium, as measured through crystal violet staining and cell counts. Values are mean ± SEM of three independent experiments in triplicates.
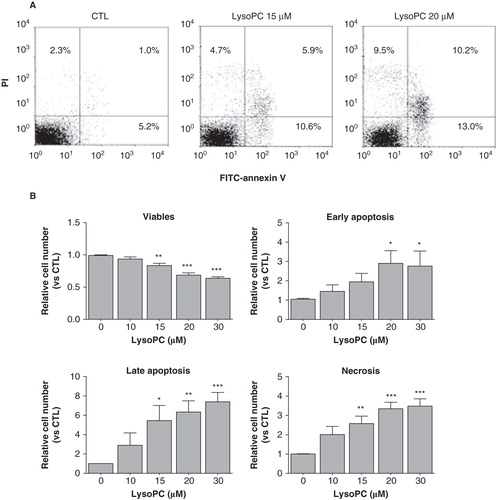
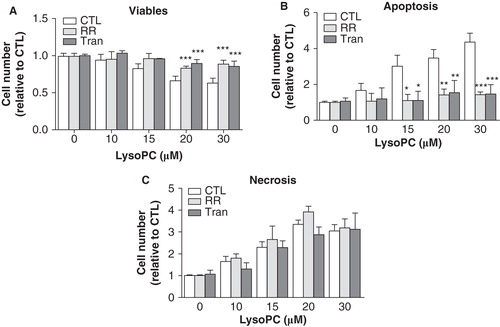
To further document lysoPC-induced cell death, we determined the levels of apoptosis and necrosis by staining adherent cells with FITC-annexin V and PI. As shown in , analysis of adherent cells after an incubation period of 24 h showed that incubation with lysoPC resulted in the appearance of cell populations that were positive for FITC-annexin V and/or positive for PI staining. Apoptosis (early apoptosis in low right quadrant and late apoptosis in up right quadrant) and necrosis (up left quadrant) were enhanced with increasing concentrations of lysoPC () which indicate that lysoPC promotes cellular apoptosis and necrosis. Mean values from adherent cells for FITC-annexin V staining (combined early and late apoptosis) were of 16% and 28% for lysoPC 15 and 30 μM, respectively, and of 12% and 22% for necrosis at 15 and 30 μM lysoPC, respectively.
Figure 2. LysoPC promotes cell death by apoptosis and necrosis. MG-63 cells seeded in 12-well plates were incubated in serum-free culture medium without lysoPC (CTL) or in the presence of increasing concentrations of lysoPC for 24 h. (A) Adherent cells were then labeled with FITC-annexin-V and propidium iodide (PI) and fluorescence was analyzed by flow cytometry. (B) Percentage of cell number relative to the control without lysoPC are shown as a function of increasing concentrations of lysoPC for viable cells, early apoptosis, late apoptosis and necrosis. Values are mean ± SEM of seven independent experiments compared to control without lysoPC using a Dunnett test: *p < 0.05, **p < 0.01, ***p < 0.001
LysoPC triggers intracellular calcium mobilization and influx
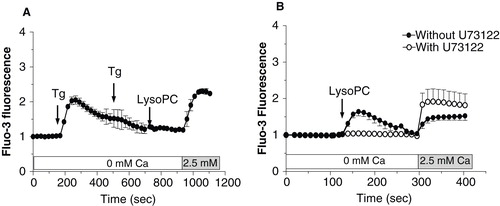
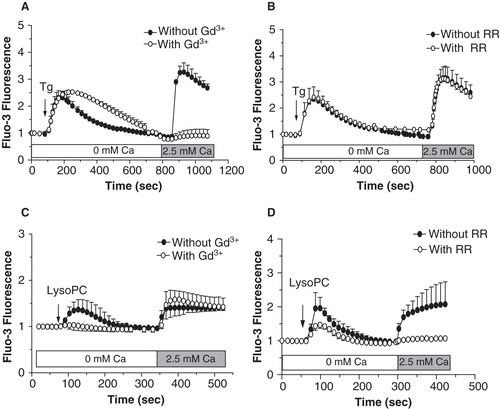
Since impairment of intracellular calcium homeostasis associated with calcium influx is often seen as a mechanism triggering cell death, we determined the involvement of extracellular calcium in the lysoPC-induced cell death. As shown in , decreasing the calcium level in the culture medium to 0.25 mM (this concentration did no compromise cell viability over 48 h) reduced the cell death induced by lysoPC suggesting that activation of calcium channels and calcium influx are involved in lysoPC-induced cytotoxicity. Therefore, we further investigated the effect of lysoPC on calcium mobilization in MG-63 cells loaded with the fluorescent calcium probe Fluo-3. Under incubation condition in calcium-free buffer, to reveal release of calcium from intracellular pools, lysoPC promoted a transient increase of intracellular calcium levels (), which correspond to a mobilization of calcium from intracellular pools. After that fluorescence had returned to the basal level, addition of calcium in the incubation buffer revealed a sustained calcium influx which increased with lysoPC concentrations. To note, no calcium influx was obtained after the addition of calcium alone without treatment with lysoPC (data not shown). Thereafter, two strategies were used to further identify origin of the intracellular calcium mobilization and determine the signaling pathway involved. First, cells were treated with 1 μM of Thapsigargine (Tg) to deplete the endoplasmic reticulum from its calcium. As shown in , calcium mobilization by lysoPC was abolished after two subsequent addition of Tg whereas the calcium influx was still observed. On the other hand, cells were pretreated with an inhibitor of PLC, namely U73122, to determine the signaling pathway triggered by lysoPC. As shown in , calcium mobilization by lysoPC was abolished in cells pretreated with U73122 whereas calcium influx was still observed. These results indicated that lysoPC induces the mobilization of calcium from the endoplasmic reticulum upon activation of PLC. To note, no calcium mobilization or influx was seen with the atherogenic lipid 7-ketocholesterol (data not shown) which has also been shown to promote cell death in MG-63 cells (Hamel et al. Citation2008).
Figure 3. Involvement of extracellular calcium in the lysoPC-induced cell death. (A) Dose-response curve of MTT activity (relative to control without lysoPC) following 48-h exposure to increasing concentrations of lysoPC in serum-free medium with low 0.25 mM calcium concentration or normal 1 mM calcium concentration. Values are mean ± SEM of five independent experiments in triplicates. Bonferroni post test: ***p < 0.001 when compared to condition with nomal 1 mM calcium. (B) Fluo-3 loaded cells were transferred to HBSS without calcium and then lysoPC (10 or 20 μM) were added to the incubation medium. Thereafter, calcium was added to the incubation buffer to determine calcium influx. Fluo-3 fluorescence values relative to basal fluorescence are showed as mean ± SEM of 3–5 experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field).
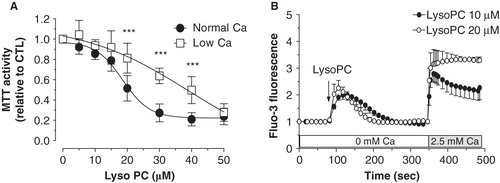
Figure 4. Characterization of calcium mobilization induced by lysoPC. (A) Cells loaded with Fluo-3 were transferred to HBSS without calcium and then calcium depletion of the endolasmic reticulum was achieved by two subsequent additions of thapsigargin (Tg 5 μM). Thereafter, lysoPC (10 μM) were added to the incubation medium. Calcium was added to the incubation buffer to determine calcium influx. (B) Cells loaded with Fluo-3 were transferred to HBSS without calcium and preincubated with 10 μM U73122 for 5 min. Thereafter, lysoPC (10 μM) were added to the incubation medium. Measurements of intracellular calcium were performed with a laser scanning confocal as described in the Material and methods section. Thereafter, calcium was added to the incubation buffer to determine calcium influx. Values are mean ± SEM of three independent experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field).
Activation of TRP channels by lysoPC
To identify which subclass family of TRP channels were associated to the lysoPC-induced calcium influx, we first characterized two TRP channel inhibitors, namely gadolinium (Gd3+) and ruthenium red (RR). As shown in , Gd3+ efficiently inhibited calcium influx induced following Tg-mediated calcium mobilization from the endoplasmic reticulum. Under these conditions, we previously showed that Tg-induced calcium mobilization from the endoplasmic reticulum triggers the activation of store-operated calcium entry mediated by TRPC1 channels in MG-63 cells (Labelle et al. Citation2007, Abed et al. Citation2009). Therefore, Gd3+ was proven as a potent inhibitor of TRPC channels under our experimental conditions. As shown in , RR was without effect on the calcium influx associated with Tg-induced calcium mobilization which agrees with the fact that RR rather acts as a known TRPV channel inhibitor. Since our results indicated that, similarly to Tg, lysoPC also induced a calcium mobilization from the endoplasmic reticulum, we asked if the lysoPC-induced calcium influx involved the activation of TRPC channels. As shown in , pretreatment of cells with Gd3+ did not prevent the calcium influx induced by lysoPC, which excludes the activation of store-operated calcium entry by lysoPC. On the other hand, the lysoPC-induced calcium influx was prevented by pretreating cells with the inhibitor of the TRPV channels RR ().
Figure 5. Characterization of calcium influx induced by lysoPC. Cells loaded with Fluo-3 were transferred to HBSS without calcium and preincubated with 100 μM Gd3+ (A, C) or 10 μM RR (B, D) for 5 min. Thereafter, 5 μM Tg (A, B) or 10 μM lysoPC (C, D) were added to the incubation medium. Calcium was added to the incubation buffer to determine calcium influx. Values are mean ± SEM of three individual experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field).
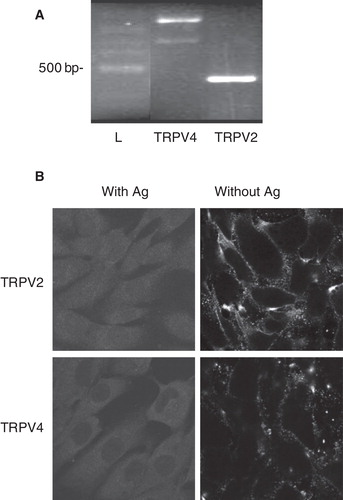
Expression of TRPV channels by MG-63 cells
We previously reported that numerous osteoblast cell lines express TRPV channels (Abed et al. Citation2009). However, the presence of corresponding proteins is still to be confirmed. Therefore, we investigated expression of TRPV in MG-63 cells. As shown in , a single PCR product was obtained for TRPV2 and two bands were observed for TRPV4 with RNA from MG-63 cells. PCR band of 686 bp corresponds to variants 2 and 3 of TRPV4 (GI294459960 and GI294459976, respectively) and amplified DNA of 866 bp corresponds to TRPV4 variants 1, 4 and 5 (GI294459961, GI294459970 and GI294459964, respectively). No PCR products were observed for the other members of TRPV family such as TRPV1, TRPV3, TRPV5 and TRPV6 (data not shown). By immunofluorescence, we confirmed the presence of TRPV2 and TRPV4 proteins with detection mainly at the plasma membrane of cells (). As our results indicate plasma membrane localization of TRPV2 and TRPV4 proteins in MG-63 cells, these channels were considered likely candidate for lysoPC-induced calcium influx.
Figure 6. Expression of TRPV channels by MG-63 cells. (A) Total RNA was isolated from MG-63 and used for synthesis of complementary DNA. Amplifications by PCR were performed with specific primers for human TRPV2 or TRPV4. Representative data are shown from RNA isolations of at least three independent cultures with amplified DNA of 686 and 866 bp for TRPV4 (corresponding to variants 2 and 3, and to variants 1, 4 and 5, respectively) and of 432 bp for TRPV2. L: ladder of 100 bp. (B) Localization of TRPV2 and TRPV4 proteins were performed by confocal microscopy with specific antibodies, and revealed by secondary fluorescence antibodies. To ascertain specific fluorescence, primary antibodies have been incubated with related antigens (Ag). Representative data are shown from at least three independent cultures.
Activation of TRPV channels by lysoPC is associated with cell death
Since our results indicated that lysoPC promotes the activation of TRPV channels and that calcium was involved in lysoPC-induced MG-63 cell death, we further characterized which TRPV channels were associated to cell death induced by lysoPC. As shown in , the decrease of MTT activity induced by lysoPC was reduced in the presence of the TRPV channel inhibitor RR and of the TRPV2 inhibitor Tranilast whereas the TRPV4 inhibitor RN 1734 was without effect. To ascertain the efficiency of RN 1734 as an inhibitor of TRPV4, we determined its inhibition potential using GSK, a known activator of TRPV4. As shown in , GSK promoted a calcium influx in MG-63 cells which is prevented by pretreating cells with RN 1734. Interestingly, the activation of TRPV4 channels by GSK has no effect on MTT activity (). As shown in , preincubation of cells with Tranilast significantly reduced the calcium influx induced by lysoPC to a similar level obtained with RR. Moreover, the TRPV2 activator cannabidiol (Qin et al. Citation2008) induced calcium influx () and promoted cell death in a dose-dependent manner (). Accordingly, RR and Tranilast were shown to reduce lysoPC-induced cell apoptosis but were without effect on necrosis ().
Figure 7. Involvement of TRPV activation in lysoPC-induced cell death of MG-63 cells. (A) Dose-response curve of MTT activity (relative to control without lysoPC) following 48-h exposure to increasing concentrations of lysoPC in culture medium alone (Control) or with 10 μM RR, 30 μM RN 1734 or 100 μM Tranilast (Tran). Results are from five independent experiments in triplicates. Bonferroni post test: *p < 0.05, **p < 0.01 when compared to control condition. (B) Cells loaded with Fluo-3 were transferred to fresh HBSS with 2.5 mM calcium. Thereafter, cells were preincubated for 5 min without or with 30 μM RN 1734. After, GSK1016790A (GSK, 10 nM) was added to the incubation medium and fluorescence was measured. Values are mean ± SEM of the relative fluorescence compared to basal level of five individual experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field). (C) Dose-response curve of MTT activity (relative to control without GSK) following 48-h exposure to increasing concentrations of GKS1016790A in serum-free culture medium. Values are mean ± SEM of three independent experiments performed in triplicates. (D) Fluo-3 loaded cells were transferred to HBSS without calcium and then lysoPC (10 μM) were added to the incubation medium. Thereafter, calcium was added to the incubation buffer to determine calcium influx. Fluorescence values corresponding to calcium influx relative to calcium influx induced by lysoPC are showed as mean ± SEM of 3–5 experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field). Dunnett post test: **p < 0.01, ***p < 0.001 when compared to control condition with lysoPC alone. (E) Cells loaded with Fluo-3 were transferred to fresh HBSS with 2.5 mM calcium. Thereafter, cells were preincubated for 5 min without or with 10 μM RR. After, cannabidiol (Can, 10 μM) was added to the incubation medium and fluorescence was measured. Values are mean ± SEM of three individual experiments with analyses of 7–8 fields per experiment (between 10 and 20 cells per field). (F) Dose-response curve of MTT activity (relative to control without cannabidiol) following 48-h exposure to increasing concentrations of cannabidiol in serum-free culture medium. Values are mean ± SEM of four independent experiments performed in triplicates. One-way ANOVA, p < 0.0001.
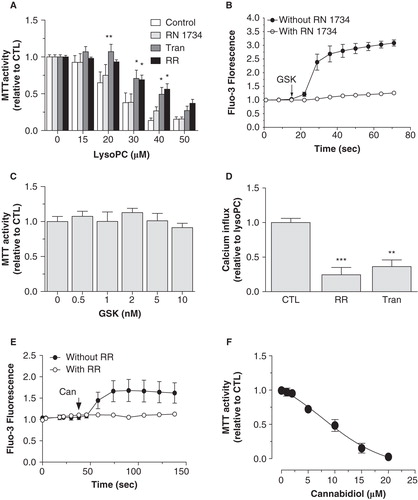
Figure 8. Stimulation of TRPV channels by lysoPC promoted cell apoptosis. MG-63 cells seeded in 12-well plates were incubated in serum-free culture medium without or with 10 μM RR or 100 μM Tranilast (Tran) in the presence of increasing concentrations of lysoPC for 24 h. The cells were then labeled with annexin-V-FITC and propidium iodide (PI) and fluorescence was analyzed by flow cytometry for viable (negative for annexin and PI), apoptotic (positive for annexin and negative for PI) and necrotic (negative for annexin and positive for PI) cells. Values are mean ± SEM of nine independent experiments. Bonferroni post test: *p < 0.05, **p < 0.01, ***p < 0.001 when compared to condition without RR or Tranilast.
Discussion
Epidemiologic and in vitro studies argue for the involvement of atherogenic conditions and OxLDL in the development of osteoporosis (Parhami et al. Citation1997, Uyama et al. Citation1997, Barengolts et al. Citation1998, Mody et al. Citation2001, Klein et al. Citation2003, Citation2006, Hamel et al. Citation2008, Brodeur et al. 2008b). Since lysoPC is a major phospholipid component of OxLDL, we investigated its effect on the viability of osteoblastic cells. Our results indicate that lysoPC induced cell death of MG-63, an effect that was dependent on extracellular calcium content which suggests that cell mortality was promoted by calcium influx induced by lysoPC. Accordingly, we reported that lysoPC promoted calcium influx by the activation of TRPV2 channels. In concordance, apoptosis induced by lysoPC was reduced by the inhibition of TRPV2 channels. Our results suggest that lysoPC promotes apoptosis in MG-63 cells by the activation of TRPV2 channels.
Oxysterols such as 7-ketocholesterol and 7β-hydroxycholesterol, as well as lysoPC are found in atherosclerotic lesions (Brown and Jessup Citation1999). These components are generated upon oxidation and enzymatic modification of LDL (Parthasarathy et al. Citation2010) and are implicated as critical factors in the atherogenic activity of OxLDL (Schmitz and Ruebsaamen Citation2010) and probably for the development of osteoporosis. Our results indicate that lysoPC induced cell death of osteoblast-like MG-63 cells. Cytotoxicity was monitored by the reduction of MTT activity, crystal violet staining and cell counts, and experiments revealed a LC50 values between 16.5 and 18.7 μM. Hsu et al. (Citation2011) have reported LC50 values between 5–10 μM for cytotoxicity of lysoPC in primary culture of vascular smooth muscle cells. Other study has reported around 37 μM for the same cell type (Hsieh et al. Citation2000). Values of LC50 for lysoPC were from 50–100 μM in endothelial cells (Takahashi et al. Citation2002, Matsubara and Hasegawa Citation2005). Therefore, our results agree with the cytotoxicity of lysoPC at micromolar concentrations. Further investigations on the mode of cell death as a function of the concentrations revealed that lysoPC promoted both apoptosis and necrosis of MG-63 cells. Hsieh et al. (Citation2000) also reported induction of apoptosis and necrosis by lysoPC in vascular smooth muscle cells. Zhou et al. (Citation2006) report apoptosis and necrosis in endothelial cells. To note, lysoPC is an amphipathic molecule and in accordance, possesses ‘detergent-like' properties which have been studies extensively in relation to lysis of red blood cells (Weltzien Citation1979). Therefore, induction of necrosis by lysoPC may likely be attributed to such membrane-perturbating property.
Since dysfunctional regulation of intracellular calcium homeostasis is often associated to cell death, we determined the involvement of extracellular calcium in the lysoPC-induced cell death. LysoPC-induced cytotoxicity was reduced when cells were incubated in low calcium medium. To note, cytotoxicity induced by the atherogenic oxysterol 7-ketocholesterol was not modified by the calcium content of incubation medium (data not shown). Therefore, specific mechanism involving extracellular calcium is triggered upon induction of cell death by lysoPC. Accordingly, our results indicated that lysoPC induces PLC-mediated calcium mobilization from endoplasmic reticulum and calcium influx. Two G protein-coupled receptors for lysoPC have been proposed, namely GPR4 and G2A (Meyer zu and Jakobs Citation2007). To note, G2A shows high binding affinity for lysoPC and was shown to be coupled to Gq protein. Also, G2A was shown to promote apoptosis in the presence of lysoPC (Lin and Ye Citation2003). Therefore, our data showing PLC-mediated calcium mobilization by lysoPC would be in accordance with the involvement of G2A as the lysoPC receptor in MG-63 cells. To the best of our knowledge, no study has yet investigated the expression of G2A in osteoblasts.
In addition to promote calcium mobilization from endoplasmic reticulum, our results indicate that lysoPC stimulate a calcium influx. We previously showed gene expression of numerous TRP channels by MG-63 cells (Abed et al. Citation2009). Moreover, we have shown that Tg- and mitogen-induced calcium mobilization from endoplasmic reticulum trigger the activation of store-operated calcium entry mediated by TRPC channels in MG-63 cells (Labelle et al. Citation2007, Abed et al. Citation2009). Since, lysoPC trigger calcium mobilization from the endoplasmic reticulum, subsequent activation of TRPC channels was plausible. Accordingly, lysoPC was shown to activate TRPC6 channels in corporal smooth muscle cells (So et al. Citation2005), suggesting a mechanism linking atherogenic condition to erectile dysfunction. LysoPC was shown to activate TRPC5 channels which confer to this type of channel the property of a lipid ionotropic receptor (Flemming et al. Citation2006). LysoPC was shown to activate TRPC5 and TRPC6 which result in the inhibition of endothelial cell migration (Chaudhuri et al. Citation2008). Nevertheless, lysoPC-induced calcium influx in MG-63 cells was not prevented by gadolinium, although our results demonstrate that this inhibitor efficiently blocked Tg-induced calcium influx in MG-63 cells. Therefore, our results suggest that lysoPC activates other members of the TRP channel family. We used RR as a known inhibitor of TRPV channels. Our results showed that lysoPC-induced calcium influx was fully inhibited by RR, suggesting that lysoPC activates TRPV channels. One study has reported activation of TRPV2 channels by lysoPC in prostate cancer cells (Monet et al. Citation2009), an effect that was associated with stimulation of cell migration. Therefore, our results further evidence that lysoPC activates TRPV channels in osteoblasts. Accordingly, our results indicate that TRPV2 and TRPV4 channels were localized to the plasma membrane of MG-63 cells, where they can assure calcium influx upon activation by lysoPC. Of interest, our results indicated that GSK promotes TRPV4-mediated calcium influx in MG-63 cell and that such activation of TRPV4 by GSK had no effect of cell viability suggesting that TRPV4-mediated calcium influx is not associated to cell death in MG-63 cells. On the other hand, the known blocker of TRPV2 Tranilast was shown to inhibit calcium influx suggesting that TRPV2-mediated calcium influx was involved in the lysoPC-induced cell death. In agreement with the reduced lysoPC-induced cell death observed in low calcium medium, our results indicate that inhibition of TRPV channels by RR and inhibition of TRPV2 by Tranilast decreased the cytotoxicity of lysoPC. Moreover, inhibition of TRPV channels by RR and of TRPV2 by Tranilast reduced lysoPC-induced apoptosis of MG-63 cells whereas being without effect on necrosis. Our data agree with previous studies which indicate that intracellular calcium signaling was associated to lysoPC-induced apoptosis of smooth muscle and endothelial cells (Zhou et al. Citation2006, Song et al. Citation2010, Hsu et al. Citation2011). In contrast to apoptosis which is a tightly regulated process involving activation and enzymatic steps with maintenance of plasma membrane integrity, necrosis is associated to plasma membrane disruption and impairment of homeostasis. Therefore, the absence of effect of RR and Tranilast on lysoPC-induced necrosis agrees with necrosis being a disorganized process.
Our results indicate that the specific activation of TRPV2 channels and related calcium influx are involved in lysoPC-induced MG-63 cell death. Intracellular calcium is a common signaling mechanism which regulates numerous cell functions such as contraction, migration, proliferation, secretion and apoptosis. Since it regulates numbers of cell functions as a second messenger, the intracellular calcium level is tightly regulated in term of amplitude, duration and subcellular localization. Although numerous types of calcium channels are expressed in the plasma membrane and although all promote calcium influx, their intrinsic properties will result in distinct calcium influx which will result in specific signaling responses and specific modulation of cell functions. In contrast to TRPV4 channels that are mainly associated with transient calcium influx due to inactivation and desensitization by increased intracellular calcium levels (reviewed in Nilius et al. Citation2004), the activity of TRPV2 seems less sensitive to intracellular calcium levels (Phelps et al. Citation2010), allowing thus a more sustained influx of calcium. In accordance, TRPV2 has been associated with a calcium-overloading pathway leading to dystrophic cardiomyopathy (Iwata et al. Citation2003). Therefore, such intrinsic channel characteristic of TRPV2 may underlay the association of TRPV2 activation with lysoPC-induced MG-63 cell death.
Altogether, our results show that lysoPC induces cytotoxicity in MG-63 cells by activating TRPV2 and our data are in accordance with the role of lysoPC as an atherogenic factor contributing to the development of osteoporosis.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC, RM).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abed E, Labelle D, Martineau C, Loghin A, Moreau R. 2009. Expression of transient receptor potential (TRP) channels in human and murine osteoblast-like cells. Mol Membr Biol 26:146–158.
- Abed E, Moreau R. 2007. Importance of melastatin-like transient receptor potential 7 and cations (magnesium, calcium) in human osteoblast-like cell proliferation. Cell Prolif 40:849–865.
- Barengolts EI, Berman M, Kukreja SC, Kouznetsova T, Lin C, Chomka EV. 1998. Osteoporosis and coronary atherosclerosis in asymptomatic postmenopausal women. Calcif Tissue Int 62:209–213.
- Brodeur MR, Brissette L, Falstrault L, Luangrath V, Moreau R. 2008a. Scavenger receptor of class B expressed by osteoblastic cells are implicated in the uptake of cholesteryl ester and estradiol from LDL and HDL3. J Bone Miner Res 23:326–337.
- Brodeur MR, Brissette L, Falstrault L, Ouellet P, Moreau R. 2008b. Influence of oxidized low-density lipoproteins (LDL) on the viability of osteoblastic cells. Free Radic Biol Med 44:506–517.
- Brown AJ, Jessup W. 1999. Oxysterols and atherosclerosis. Atherosclerosis 142:1–28.
- Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, et al. 1977. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation 55:767–772.
- Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. 2008. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell 19:3203–3211.
- Flemming PK, Dedman AM, Xu SZ, Li J, Zeng F, Naylor J, et al. 2006. Sensing of lysophospholipids by TRPC5 calcium channel. J Biol Chem 281:4977–4982.
- Fuchs B, Schiller J. 2009. Lysophospholipids: Their generation, physiological role and detection. Are they important disease markers? Mini Rev Med Chem 9:368–378.
- Hamel P, Abed E, Brissette L, Moreau R. 2008. Characterization of oxidized low density lipoproteins-induced hormesis-like effects in osteoblastic cells. Am J Physiol Cell Physiol 294:C1021–C1033.
- Hsieh CC, Yen MH, Liu HW, Lau YT. 2000. Lysophosphatidylcholine induces apoptotic and non-apoptotic death in vascular smooth muscle cells: In comparison with oxidized LDL. Atherosclerosis 151:481–491.
- Hsu JH, Wu JR, Liou SF, Chen HM, Dai ZK, Chen IJ, et al. 2011. Labedipinedilol-A prevents lysophosphatidylcholine-induced vascular smooth muscle cell death through reducing reactive oxygen species production and anti-apoptosis. Atherosclerosis 217:379–386.
- Ingueneau C, Huynh UD, Marcheix B, Athias A, Gambert P, Negre-Salvayre A, et al. 2009. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J Cell Mol Med 13:1620–1631.
- Iwata Y, Katanosaka Y, Arai Y, Komamura K, Miyatake K, Shigekawa M. 2003. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J Cell Biol 161:957–967.
- Klein BY, Kerem Z, Rojansky N. 2006. LDL induces Saos2 osteoblasts death via Akt pathways responsive to a neutral sphingomyelinase inhibitor. J Cell Biochem 98:661–671.
- Klein BY, Rojansky N, Ben Yehuda A, Abou-Atta I, Abedat S, Friedman G. 2003. Cell death in cultured human Saos2 osteoblasts exposed to low-density lipoprotein. J Cell Biochem 90:42–58.
- Labelle D, Jumarie C, Moreau R. 2007. Capacitative calcium entry and proliferation of human osteoblast-like MG-63 cells. Cell Prolif 40:866–884.
- Lin P, Ye RD. 2003. The lysophospholipid receptor G2A activates a specific combination of G proteins and promotes apoptosis. J Biol Chem 278:14379–14386.
- Matsubara M, Hasegawa K. 2005. Benidipine, a dihydropyridine- calcium channel blocker, prevents lysophosphatidylcholine-induced injury and reactive oxygen species production in human aortic endothelial cells. Atherosclerosis 178:57–66.
- Meyer zu HD, Jakobs KH. 2007. Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta 1768:923–940.
- Miller BA. 2006. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol 209:31–41.
- Mody N, Parhami F, Sarafian TA, Demer LL. 2001. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med 31:509–519.
- Monet M, Gkika D, Lehen'kyi V, Pourtier A, Vanden Abeele F, Bidaux G, et al. 2009. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim Biophys Acta 1793:528–539.
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. 2004. TRPV4 calcium entry channel: A paradigm for gating diversity. Am J Physiol Cell Physiol 286:C195–C205.
- Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, et al. 1997. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol 17: 680–687.
- Parhami F, Tintut Y, Beamer WG, Gharavi N, Goodman W, Demer LL. 2001. Atherogenic high-fat diet reduces bone mineralization in mice. J Bone Miner Res 16:182–188.
- Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N. 2010. Oxidized low-density lipoprotein. Methods Mol Biol 610:403–417.
- Pedersen SF, Owsianik G, Nilius B. 2005. TRP channels: An overview. Cell Calcium 38:233–252.
- Phelps CB, Wang RR, Choo SS, Gaudet R. 2010. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem 285:731–740.
- Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. 2008. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci 28:6231–6238.
- Schmitz G, Ruebsaamen K. 2010. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis 208:10–18.
- So I, Chae MR, Kim SJ, Lee SW. 2005. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces the change of calcium mobilization via TRPC ion channels in cultured human corporal smooth muscle cells. Int J Impot Res 17:475–483.
- Song J, Liu K, Yi J, Zhu D, Liu G, Liu B. 2010. Luteolin inhibits lysophosphatidylcholine-induced apoptosis in endothelial cells by a calcium/mitocondrion/caspases-dependent pathway. Planta Med 76:433–438.
- Steinberg D. 1997. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem 272:20963–20966.
- Takahashi M, Okazaki H, Ogata Y, Takeuchi K, Ikeda U, Shimada K. 2002. Lysophosphatidylcholine induces apoptosis in human endothelial cells through a p38-mitogen-activated protein kinase-dependent mechanism. Atherosclerosis 161:387–394.
- Uyama O, Yoshimoto Y, Yamamoto Y, Kawai A. 1997. Bone changes and carotid atherosclerosis in postmenopausal women. Stroke 28:1730–1732.
- Weltzien HU. 1979. Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim Biophys Acta 559: 259–287.
- Zhou L, Shi M, Guo Z, Brisbon W, Hoover R, Yang H. 2006. Different cytotoxic injuries induced by lysophosphatidylcholine and 7-ketocholesterol in mouse endothelial cells. Endothelium 13:213–226.