Abstract
Recent advances have revealed much about the signaling events and molecular components associated with autophagy. Although it is clear that there are multiple points of intersection and connection between autophagy and known vesicular membrane transport processes between membrane compartments, autophagy is modulated by a distinct set of molecular components, and the autophagosome has a unique membrane composition. A key question that has yet to be resolved with regards to autophagosome formation is its membrane source. Various evidences have indicated that membranes from the endoplasmic reticulum (ER), mitochondria, Golgi, endosomes and the plasma membrane could all potentially act as a source of autophagosomal membrane in non-specialized macroautophagy. Recent investigations have generated advances in terms of the mitochondria's involvement in starvation-induced autophagy, and refined localization of autophagosome formation to ER-mitochondria contact sites. On the other hand, data accumulates on the delivery of membrane sources to the pre-autophagosome structure by Atg9-containing mobile carriers, which likely originated from the Golgi-endosome system. The answer to the question on the origin of membrane materials for autophagosome biogenesis awaits further reconciliation of these different findings.
Introduction
Macroautophagy is a general term describing cellular processes by which cytoplasmic content and membranous organelles are non-specifically engulfed by a double-membrane autophagosome, and then degraded at the vacuole (in yeast) or lysosome (in vertebrates) (Yang and Klionsky 2010, Tanida Citation2011, Rubinsztein et al. Citation2012). More specific autophagic processes involving particular organelles have also been described for contents of the ER (reticulophagy, (Cebollero et al. Citation2012)), mitochondria (mitophagy, (Ding and Yin Citation2012)) and peroxisomes (pexophagy, (Manjithaya et al. Citation2010)). Two other forms of cellular degradation, namely microautophagy (Li et al. Citation2012) and chaperone-mediated autophagy (Kaushik and Cuervo Citation2012), should be distinguished from the non-selective process of macroautophagy (which is hereafter simply referred to as ‘autophagy'). Autophagy plays critical roles in cellular homeostasis during development and postnatal survival. Impairment of autophagic activities is increasingly associated with disease conditions such as cancer (Rosenfeldt and Ryan Citation2011), neurodegeneration (Banerjee et al. Citation2010) metabolic/cardiovascular disorders (Ouimet Citation2013), as well as organismal aging (Gelino and Hansen Citation2012).
Autophagy is initiated by a network of molecular complexes modulating autophagosome formation and maturation. Our knowledge of the molecular machineries involved in the process centers round the set of Autophagy (Atg) related proteins whose genes are first identified by genetic screens in yeast (Reggiori and Klionsky Citation2002). The Atg proteins function in various complexes which together, in a complicated and apparently non-linear manner, generate the autophagosome. What is now known about this process has been extensively reviewed elsewhere (Simonsen and Tooze Citation2009, Yang and Klionsky 2010, Klionsky Citation2013), and will only be very briefly described below. Autophagy could be induced by cellular stress or nutrient starvation, and these are often used experimental paradigms to probe autophagic processes in cellular experimental models. The Atg1/unc51-like kinase 1 (ULK1) complex (Wong et al. Citation2013), containing Atg1/ULK1, Atg17, and dephosphorylated Atg13 is an upstream effector of autophagic signalling connected to the mechanistic target of rapamycin (mTOR) (Zoncu et al. Citation2011) and AMP-activated kinase (AMPK) modulated nutrient-sensing pathways (Yuan et al. Citation2013). This Atg1/ULK1 complex appears to act in recruiting other Atg proteins to the autophagosome assembly sites (Reggiori et al. Citation2004, Cheong et al. Citation2008, Sekito et al. Citation2009) to initiate the formation of the pre-autophagosmal membrane/phagophore (or the mammalian isolation membrane [IM]) (Hayashi-Nishino et al. Citation2009). Another protein complex, containing the class III phosphatidylinositol-3 (PI3)-kinase Vps34, Atg6/beclin1, Vps15/p150 and Atg14/Atg14L, functions at the onset of autophagy through local production of phosphatidylinositol 3-phosphate (PI3P) (Axe et al. Citation2008, Walker et al. Citation2008), which is enriched in the inner surface of the IM, and marked by the double FYVE domain-containing protein 1 (DFCP1), a PI3P-binding protein (Axe et al. Citation2008). A mechanistic link between the two complexes has been established by a recent description of ULK1 inducing autophagy by phosphorylating Beclin-1 and activating the Vps34 lipid kinase (Russell et al. Citation2013). This PI3P in turn engages scaffold proteins such as the yeast Atg18 and Atg21 (or their mammalian homologues WD-40 repeat containing protein that Interacts with Phosphatidyinositols [WIPI] 1 and 2) (Polson et al. Citation2010), and becomes a membrane platform for accumulation of autophagosomal proteins and subsequent IM growth and maturation.
The subsequent expansion and eventual closure of the IM to form a free autophagic vesicle is dependent on two ubiquitin-like conjugation processes, leading to the formation of conjugates with membrane phosphatidylethanolamine (PE) (Noda et al. Citation2008). The E1-like Atg7 and E2-like Atg10 catalyze the conjugation of the ubiquitin-like protein Atg12 to Atg5, and facilitate the subsequent formation of the Atg16L-containing complex (Fujita et al. Citation2008). This Atg5–Atg12–Atg16L complex is localized to the IM, and acts like an E3-ligase in promoting the recruitment of the other ubiquitin-like protein Atg8/microtubule-associated protein 1A/1B light chain 3 (MAP-LC3, or simply LC3, the cytoplasmic form is also designated as LC3-I) to PE on the membrane (Fujita et al. Citation2008). LC3-phosphatidylethanolamine conjugate (LC3-II) on the membrane facilitates autophagosome formation, presumably by enabling membrane elongation. Indeed, LC3 rather specifically decorates autophagosomal membranes, and detection of cytoplasmic and membrane-associated LC3 (or GFP-LC3) is a simple and popular method for monitoring autophagy progression (Tanida et al. Citation2008). Atg8 also binds Atg1/Ulk1, an interaction that may somehow promote autophagosome maturation and lysosomal fusion (Kraft et al. Citation2012). Another component known to be involved in IM expansion is Bax-interacting factor 1 (Bif-1)/Endophilin B1 (Takahashi et al. Citation2009). Bif-1 forms a complex with Atg6/beclin1, and may act in driving membrane curvature in the processes of IM extension and closure (Takahashi et al. Citation2009). In spite of the increasing insight into the molecular components mediating autophagosome formation, two key questions have remained unclear.
The questions: Where do autophagosomes form and what is the source(s) of autophagosomal membrane?
Details concerning the initial steps of IM formation have been intensively investigated, and debated. The first question appears to be easy to answer. In yeast, autophagosome assembly typically begins at a single perivacuolar site (morphologically a single spot near the vacuole) called the phagophore assembly site (PAS) (Cheong et al. Citation2008). Mammalian cells appear to have multiple autophagosome assembly sites (Hamasaki and Yoshimori Citation2010, Tooze and Yoshimori Citation2010) characterized by the isolation membrane (IM), which appears larger than the PAS of yeast. One of the key questions concerns the source of membrane material that forms the IM. In many morphological studies, the PAS or IM appears intimately associated with, or physically connected to, the ER (Hayashi-Nishino et al. Citation2009, Ylä-Anttila et al. Citation2009). Functionally, autophagy is known to require ER-targeting of the PI3-kinase complex via Atg14/Atg14L (Matsunaga et al. Citation2010). However, isolated autophagosomes lack ER markers and appear to be rather unique in terms of composition (Strømhaug et al. Citation1998). It has also been shown that the mitochondria are a major membrane source for autophagosome formation (Hailey et al. Citation2010). A more recent finding provided some reconciliation of the above by showing that autophagosomes form from ER-mitochondria contact sites (Hamasaki et al. Citation2013), which are the connective interfaces between the two organelles with known functions in regulating calcium signaling, lipid synthesis and mitochondrial biogenesis (Rowland and Voeltz Citation2012).
The only multipass transmembrane protein amongst the Atg proteins that is required for optimal autophagy is Atg9, and one would intuitively feel that following its dynamics in the cell upon induction of autophagy would be revealing as far as an autophagosome membrane source(s) is concerned. Indeed, in yeast Atg9's Atg11-dependent recruitment to PAS is critical for autophagy (He et al. Citation2006), and mobile Atg9-containing vesicles have been shown to be a membrane source for autophagosome formation in yeast. However, morphological studies did not provide a clear consensus as to where these Atg9 containing vesicles come from. Atg9 has been shown to concentrate in a ‘novel' compartment comprising clusters of vesicles and tubules that are often adjacent to the mitochondria (Mari et al. Citation2010). Another study with a combination of yeast mutants indicated that Atg9 is unlikely to be delivered to PAS directly from the mitochondria or ER, but more likely from the Golgi-endosomal membranes (Ohashi and Munro Citation2010). More recent work in yeast has also demonstrated that much of cellular Atg9 resides on cytoplasmic mobile vesicles that were derived from the Golgi apparatus (Yamamoto et al. Citation2012). The mammalian homologue of Atg9, mAtg9, shown to be localized to the trans-Golgi network, late endosomes and recycling endosomes (Young et al. Citation2006, Orsi et al. Citation2012), is required for the formation of phagophores marked by DFCP1 (Orsi et al. Citation2012).
The recent findings, while interesting and insightful, provided no apparent morphological or functional connection between the pre-autophagosome assembly site and its membrane source. Below, we describe and discuss these recent findings in more detail.
Association and connection of autophagosome formation sites and the isolation membrane with the ER
Firm support for the ER, or a subdomain of the ER, as the main site of autophagosome formation came from several notable morphological studies late in the last decade. Axe et al. (Citation2008) showed that the PI3P-binding DFCP1, a marker for autophagosome assembly sites, has an ER targeting domain and is ER-associated. Upon autophagy induction by starvation, both DFCP1 and a reporter molecule consisting of the FYVE domain anchored on the ER membrane by a transmembrane domain (GFP-FYVE-TM) translocated, in a PI3P-dependent manner, to torus ring-like punctate structures which the authors termed omegasome. The latter appears to form de novo and eventually collapses with time, is in close contiguity with, and is dynamically associated with ER membranes. Autophagosomes form within these omegasomes, and these eventually acquire LC3 coating, lose DFCP1 staining, and exit the omegasome. The data illustrated the formation of a sort of ER-associated membrane cradle that forms the autophagosome. Another key support for a role of ER as a platform in autophagosome formation in mammalian cells came from the demonstration that the PI3 kinase complex required for the generation of PI3P is targeted to the ER by Atg14L, and silencing or mutation of Atg14L both diminished the DFCP1-positive omegasomes (Matsunaga et al. Citation2010).
The morphology of this ER-based membrane cradle was explored at the ultrastructural level by two reports using electron microcopy (EM) tomography (Hayashi-Nishino et al. Citation2009, Ylä-Anttila et al. Citation2009). Hayashi-Nishino and colleagues studied the structure of forming IMs by overexpressing an inactive mutant of Atg4B, which inhibits conversion of LC3-I to LC3-II and causes accumulation of defective, unclosed autophagosomal membrane (Hayashi-Nishino et al. Citation2009). The authors observed by EM a subdomain of the ER forming a cradle-like curve encircling the IM. The IM itself appears to also enclose an ER cisterna and thus appeared sandwiched between two cisternae. Intriguingly, vesicular clusters were visible at the vicinity of the IM, and these structures morphologically resembled the vesicular tubular clusters (VTC), which are COPII-mediated ER exit sites for secretory transport (Bannykh et al. Citation1996). Importantly, the authors showed that the associated ER and IM were connected by a membrane extension from the IM. Likewise, 3D tomographic imaging by Ylä-Anttila et al. (Citation2009) showed connections between the IM and the closely located ER cisternae.
The studies summarized above strongly suggest that although the IM did not directly originate from the ER, the latter appears pivotal for its formation. An observed membranous connection by EM, however, says little about actual membrane flux between the two compartments. A stronger hint of a dynamic connection between the ER and the IM came from another recent finding that the scaffold protein Atg11, which interacts with a few other Atg proteins, is an effector of Rab1/Ypt1, the small GTPase regulating ER-Golgi transport (Jedd et al. Citation1995). Segev and colleagues showed that Ypt1-Atg11 interaction is required for PAS formation, and involves Ypt1 activation (GDP-GTP exchange) by a unique Transport protein particle III (TRAPP III) complex containing a Trs85 subunit (Lipatova et al. Citation2012, Lipatova and Segev Citation2012). Interestingly, the authors also show that Ypt1 and Trs85 interact on Atg9-containing membranes, which, as mentioned above and shall be discussed further below, are recognized sources for the PAS membrane. It was shown earlier that Atg11 recruits Atg9 to pre-autophagosomes and this recruitment is essential for autophagy (He et al. Citation2006). More recently, it was found that Trs85 directly interacts with Atg9, and Trs85-containing TRAPP III facilitates the recruitment of Ypt1 to the Atg9-containing vesicles (Kakuta et al. Citation2012). It is therefore conceivable that Ypt1's engagement of Atg11 may divert membrane flux from ER-Golgi to ER-IM (see part D of ), although this simplistic notion lacks any clear evidence just as yet.
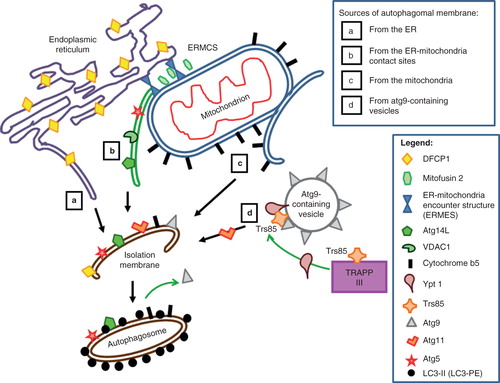
Figure 1. Possible membrane sources for forming autophagosome, or isolation membrane (IM), and the various autophagy (Atg) components and other markers associated with these. Four possible sources have been proposed to give rise to the IM. (a) Autophagosomal membrane from the endoplasmic reticulum. DFCP1, which marks the autophagosome assembly sites, is also ER-associated. (b) IM contribution from the ER-mitochondria contact site (ERMCS). Protein markers shown on this IM include the mitochondria-associated ER membrane (MAM)-enriched voltage-dependent anion channel 1 (VDAC1) and autophagy proteins Atg5 and Atg14L. The contact site is maintained by tethering molecules such as the ER-mitochondria encounter structure (ERMES) complex (Kornmann et al. Citation2009). (c) IM contribution from the mitochondria outer membrane. A mitochondrial outer membrane protein marker cytochrome b5 (cb5) is present on autophagosomes induced by starvation. (d) IM contribution from Atg9-containing vesicles. Markers such as Ypt1 and Trs85 interact on the membranes of these vesicles. Atg11, together with Ypt1, facilitates pre-autophagosome formation and recruits Atg9 to the pre-autophagosomes.
Mitochondrial membrane involvement in starvation-induced autophagy
Entire mitochondria could be degraded by a specific autophagic process, namely mitophagy (Ding and Yin Citation2012). However, in a very elegant study, Hailey et al. (Citation2010) demonstrated that the mitochondrial outer membrane, but not the ER membrane, contributes membrane and lipids to the forming autophagosome. The authors investigated the overlap of YFP-tagged membrane targeting markers with CFP-LC3 and found that only the mitochondrial outer membrane marker based on mitochondrial cytochrome b5 (cb5), YFP-Mitocb5TM, uniquely co-localized with LC3 after starvation-induced autophagy. This occurs uniquely with autophagosomes formed during starvation, and not autophagosomes induced by ER stress or calcium influx. Importantly, unlike mitophagy, mitochondrial matrix and inner membrane markers did not show significant LC3 colocalization. YFP-Mitocb5TM in autophagosomes was also present on the limiting membranes of autophagosomes rather than the autophagosome lumens. The authors could visualize outgrowth of autophagosomes from the mitochondria, and used Fluorescence recovery after photobleaching (FRAP) of YFP-Mitocb5TM to demonstrate membrane continuity between the mitochondrial outer membrane and the newly formed autophagosome.
An important aspect of this study is that the authors were able to demonstrate lipid delivery from the mitochondria to newly formed autophagosomes. Another key finding is the involvement of the ER-mitochondria contacts in this process. A cell line lacking mitofusin 2, a mitochondrial fusion factor shown to help tether mitochondria to ER, (de Brito and Scorrano Citation2008) could not form starvation-induced autophagosomes. Interestingly, unlike the tagged cb5, other protein markers of the outer mitochondrial membrane, such as Tom20, are also excluded from the autophagosome. The unique features of the transmembrane domain (TM) of cb5 appear to allow the protein to span only the outer leaflet of the outer membrane. A cb5 mutant that would span both leaflets of the membrane is also excluded from the autophagosome, and appending the cb5 TM could target otherwise excluded markers to the autophagosome upon starvation. The study is also important as it provided a first insight into a possible protein diffusion barrier between the forming IM and other membranes (Hailey et al. Citation2010), which would explain why autophagosomes have a unique membrane composition that excludes most other organelle markers.
If the study by Hailey et al. hinted at the involvement of ER-mitochondria contact sites in autophagosome formation, another recent study provided further confirmation for this notion. Hamasaki et al. showed that ER-mitochondria contact sites are the primary locale for autophagosome formation in mammalian cells (Hamasaki et al. Citation2013). The mitochondria-associated ER membrane (MAM) (Raturi and Simmen Citation2013), which include the contact sites, has unique biophysical and biochemical properties, and could be isolated by Percoll gradient centrifugation. The authors found that Atg14L-positive punctae assemble upon autophagy induction by starvation, and GFP-DFCP1 is also translocated there. Starvation also induced MAM association of Atg5, which marks specifically the IM, colocalizing with the MAM-enriched voltage-dependent anion channel 1(VDAC1). Silencing of the phosphofurin acidic cluster sorting protein-2 (PACS-2) previously implicated in contact site formation, also decreased LC-II accumulation and Atg14L/DFCP1 localization to MAM.
Another interesting finding made by the authors is the involvement of an ER-localized soluble N-ethylmaleimide sensitive protein attachment protein receptor (SNARE), Stx17 (Muppirala et al. Citation2011), in targeting Atg14L to the MAM upon starvation. Stx17 was previously implicated in a late step of autophagy, namely autophagosome-endosome/lysosome fusion (Itakura et al. Citation2012). Silencing of Stx17 significantly decreased autophagic flux, and tellingly resulted in an accumulation of IMs and a decrease in mature autophagosomes. Involvement of a SNARE would suggest that targeting of material to the forming autopagosome at the MAM requires membrane fusion, and that Stx17 may act in a cognate SNARE complex (Hay Citation2001) whose component SNAREs have not yet been fully identified. In this regard, however, the nature of the membrane carriers involved and the subsequent mode of fusion at MAM remained unclear. Information of what might constitute carriers came from another line of study that centers round a transmembrane protein, Atg9 (see section below). That the MAM is involved in autophagy was also implicated by other findings. The small GTPase Rab32, often implicated in the biosynthesis of melanosome and other lysosome-related organelles (Bultema and Di Pietro Citation2013), could also be localized to the ER (Hirota and Tanaka Citation2009) and the MAM (Bui et al. Citation2010), and appears to be required for autophagy in mammalian cells under basal, non-staved conditions (Hirota and Tanaka Citation2009). Rab32 in Drosophila has also been shown to regulate autophagy and the size of lipid droplets (Wang et al. Citation2012). The exact roles played by Rab32 in autophagy is not yet clear, but if known functions of Rabs are anything to go by, Rab32 may be important for vesicle trafficking to and/or vesicle docking/fusion at the MAM. In fact, several classical components of vesicular membrane traffic such as Rab proteins (Hirota and Tanaka Citation2009, Lynch-Day et al. Citation2010, Kakuta et al. Citation2012), SNAREs (Hamasaki et al. Citation2013) and Rab-dependent tether (Backues and Klionsky Citation2012) have now all been implicated in autophagosome formation. This is suggestive of a membrane-based delivery process that is mechanistically akin to that in conventional membrane traffic.
Autophagosome formation with Atg9-containing vesicles generated from the Golgi–endosome
Atg9, with six membrane spanning domains, is the only multipass transmembrane protein amongst the Atg proteins that is required for optimal autophagy (He et al. Citation2006, Young et al. Citation2006). Early work in yeast suggested that it is important for both autophagy and the cytoplasm-to-vacuole (cvt) pathway (Lang et al. Citation2000). Indeed, in yeast Atg9's recruitment by Atg11 to PAS is critical for the onset of autophagy (He et al. Citation2006). Another transmembrane protein, Atg27, is required for Atg9 cycling and itself shuttles between PAS, mitochondria, and the Golgi complex (Yen et al. Citation2007). Another report indicated that Atg9 could also be recruited to PAS by Atg17 (Sekito et al. Citation2009). So where exactly does Atg9 come from? Atg9 has been shown to shuttle between PAS and mitochondria in yeast (Reggiori et al. Citation2005). A more recent study has shown that Atg9 is concentrated in a yeast compartment comprising clusters of vesicles and tubules that are often adjacent to the mitochondria (Mari et al. Citation2010). However, another study which examined the molecular requirements for delivery of Atg9-containing membrane to the yeast autophagosome indicated that Atg9 is unlikely to be delivered to PAS from the mitochondria, or an early secretory pathway compartment such as the ER, but more likely from the Golgi-endosomal membranes (Ohashi and Munro Citation2010). Rubinsztein and colleagues have shown that the plasma membrane could be a potential source of at least some types of autophagosomes characterized by Atg16L1-positive ‘early autophagosome precursors' formed by clathrin-dependent endocytic mechanism (Ravikumar et al. Citation2010). A very recent report from the same laboratory showed that Atg9-containing vesicles are also generated in a clathrin-dependent manner, but are routed to the recycling endosomes differently from ATG16L1-containing vesicles. These carriers fuse at the recycling endosome, and the fusions were enhanced by starvation and correlated with autophagosome formation (Puri et al. Citation2013).
More recent work in yeast have also demonstrated that much of cellular Atg9 reside on cytoplasmic mobile vesicles (Yamamoto et al. Citation2012). These small (40–50 nm) single membrane vesicles are likely derived from the Golgi apparatus, as Atg9 accumulated at the TGN in mutants of the yeast Golgi ARF-GEF sec7ts , and Atg-containing vesicle generation is not impaired in mutants defective in endosome-Golgi retrograde trafficking. During autophagosome formation, Atg9 is embedded in the autophagosomal outer membrane, but is likely recycled to the cytoplasm as new Atg9-bearing vesicles.
On the other hand, the mammalian homologue of Atg9, mAtg9, was localized by Tooze's group to both a perinuclear region, which corresponds to the trans-Golgi network (TGN), and a more peripheral membrane population, which was initially shown to partially colocalize with late endosomal markers (Young et al. Citation2006). A more recent study by Orsi et al. (Citation2012) has also indicated the existence of an Atg9-enriched tubular-vesicular membrane compartment in mammalian cells, which is interpreted as endosome-like as it is marked by the endosomal marker transferrin receptor. Interestingly, these authors found that although mAtg9 is required for the formation of phagophores marked by DFCP1, it does not become a permanent part of the autophagosome membrane. The mAtg9-positive structures in fact only interact dynamically with phagophores, without being stably incorporated into these. This finding bears resemblance to that seen in yeast, which indicates that Atg9-containing vesicles are indeed cargo carriers, and that Atg9 acts like a recycled cargo adaptor. This notion is supported by the finding that Atg9 binds directly to the TRAPIII subunit Trs85 (which in turn engages the GTPases Ypt1p) and recruits these into the preautophagosome structure (Kakuta et al. Citation2012).
Conclusion
A diagrammatic summary of the various perceptions of where the isolation membrane is derived from is provided in . The recent findings discussed above on the location and membrane sources for autophagosome formation have provided useful insights into our understanding of this key cellular homeostasis process. While the findings that ER-mitochondria contact sites are where autophagosomes form may resolve some morphologically puzzling discrepancies (i.e., the ‘ER origin' versus ‘mitochondria-origin' models), how exactly are membrane materials delivered to the phagophore or IM is unclear. It may be a little while more before the pieces of the puzzle come together. However, we could speculatively entertain the following notions. For starvation-induced autophagy, a mechanism of unidirectional transport mediated by the membranous connection seen between ER/mitochondria, and regulated by diffusion filters, could explain how mitochondrial lipids and selected outer membrane components could be delivered to the forming autophagosome. Atg9-bearing vesicles may carry a different set of components to the autophagosome (such as Ypt1), and it appears that multiple membrane transport processes contribute to the formation and growth of the isolation membrane.
Several important questions are left unanswered. Where exactly are the Atg9-bearing mobile carriers from, how do these form and how are these targeted to PAS or autophagosome formation site in mammalian cells? Does its generation and targeting involve a novel vesicular trafficking mechanism? What are the nature and composition of the junction between the visualized membrane contact between IM and ER or mitochondria membrane? Perhaps the most puzzling is the finding that the mitochondria membrane dependence is peculiar to starvation-induced autophagy and not stress-induced autophagy. Are the latter therefore dependent on other modes of material recruitment? One other possible connection, albeit speculative at the moment, is that autophagosome formation may stem from localized signals, such as reactive oxygen species (ROS) generated by the respective membranous structures. ER stress induces production of ROS at the ER while starvation elevates mitochondrial ROS, and mitochondrial ROS, particularly superoxide, have been shown to regulate autophagy (Chen et al. Citation2009, Li et al. Citation2013).
Furthermore, there are non-canonical autophagic processes which are apparently independent of some of the key Atg proteins (Juenemann and Reits Citation2012), such as the recently described Atg5/Atg7-independent alternative macroautophagy (Nishida et al. Citation2009). In this latter mode, autophagosomes are generated in a Rab9-dependent manner, apparent with vesicles derived from the TGN and late endosomes. These could well be the same Atg9-bearing mobile carriers, and we await further enlightenment on these issues.
Acknowledgements
The authors are supported by NUS Graduate School for Integrative Sciences and Engineering (NGS). We thank the reviewers for their constructive comments which improved the manuscript.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701.
- Backues SK, Klionsky DJ. 2012. Atg11: A Rab-dependent, coiled-coil membrane protein that acts as a tether for autophagy. Autophagy 8:1275–1278.
- Banerjee R, Beal MF, Thomas B. 2010. Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends Neurosci 33:541–549.
- Bannykh SI, Rowe T, Balch WE. 1996. The organization of endoplasmic reticulum export complexes. J Cell Biol 135:19–35.
- Bui M, Gilady SY, Fitzsimmons REB, Benson MD, Lynes EM, Gesson K, et al. 2010. Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem 285:31590–31602.
- Bultema JJ, Di Pietro SM. 2013. Cell type-specific Rab32 and Rab38 cooperate with the ubiquitous lysosome biogenesis machinery to synthesize specialized lysosome-related organelles. Small GTPases 4:16–21.
- Cebollero E, Reggiori F, Kraft C. 2012. Reticulophagy and ribophagy: Regulated degradation of protein production factories. Int J Cell Biol 2012:182834.
- Chen Y, Azad MB, Gibson SB. 2009. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16:1040–1052.
- Cheong H, Nair U, Geng J, Klionsky DJ. 2008. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell 19:668–681.
- de Brito OM, Scorrano L. 2008. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610.
- Ding WX, Yin XM. 2012. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol Chem 393:547–564.
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. 2008. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19:2092–2100.
- Gelino S, Hansen M. 2012. Autophagy – an emerging anti-aging mechanism. J Clin Experim Pathol Suppl 4.
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. 2010. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667.
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393.
- Hamasaki M, Yoshimori T. 2010. Where do they come from? Insights into autophagosome formation. FEBS Lett 584:1296–1301.
- Hay JC. 2001. SNARE complex structure and function. Exp Cell Res 271:10–21.
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. 2009. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11:1433–1437.
- He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, et al. 2006. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol 175:925–935.
- Hirota Y, Tanaka Y. 2009. A small GTPase, human Rab32, is required for the formation of autophagic vacuoles under basal conditions. Cell Mol Life Sci 66:2913–2932.
- Itakura E, Kishi-Itakura C, Mizushima N. 2012. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151:1256–1269.
- Jedd G, Richardson C, Litt R, Segev N. 1995. The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J Cell Biol 131:583–590.
- Juenemann K, Reits EA. 2012. Alternative macroautophagic pathways. Int J Cell Biol 2012:189794.
- Kakuta S, Yamamoto H, Negishi L, Kondo-Kakuta C, Hayashi N, Ohsumi Y. 2012. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J Biol Chem 287:44261–44269.
- Kaushik S, Cuervo AM. 2012. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol 22:407–417.
- Klionsky D. 2013. An overview of autophagy: Morphology, mechanism and regulation. Antioxid Redox Signal.
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, et al. 2009. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481.
- Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, et al. 2012. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J 31:3691–3703.
- Lang T, Reiche S, Straub M, Bredschneider M, Thumm M. 2000. Autophagy and the cvt pathway both depend on AUT9. J Bacteriol 182:2125–2133.
- Li L, Chen Y, Gibson SB. 2013. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 25:50–65.
- Li WW, Li J, Bao JK. 2012. Microautophagy: Lesser-known self-eating. Cell Mol Life Sci 69:1125–1136.
- Lipatova Z, Belogortseva N, Zhang XQ, Kim J, Taussig D, Segev N. 2012. Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proc Natl Acad Sci USA 109:6981–6986.
- Lipatova Z, Segev N. 2012. A Ypt/Rab GTPase module makes a PAS. Autophagy 8:1271–1272.
- Lynch-Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, et al. 2010. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci USA 107:7811–7816.
- Manjithaya R, Nazarko TY, Farré JC, Subramani S. 2010. Molecular mechanism and physiological role of pexophagy. FEBS Lett 584:1367–1373.
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. 2010. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190:1005–1022.
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. 2010. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190:511–521.
- Muppirala M, Gupta V, Swarup G. 2011. Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol Cell 103:333–350.
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. 2009. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658.
- Noda T, Fujita N, Yoshimori T. 2008. The Ubi brothers reunited. Autophagy 4:540–541.
- Ohashi Y, Munro S. 2010. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell 21:3998–4008.
- Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. 2012. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23:1860–1873.
- Ouimet M. 2013. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Biochim Biophys Acta 1831:1124–1133.
- Polson HEJ, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. 2010. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6:506–522.
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. 2013. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154:1285–1299.
- Raturi A, Simmen T. 2013. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833:213–224.
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. 2010. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12:747–757.
- Reggiori F, Klionsky DJ. 2002. Autophagy in the eukaryotic cell. Eukaryot Cell 1:11–21.
- Reggiori F, Shintani T, Nair U, Klionsky DJ. 2005. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 1:101–109.
- Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. 2004. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell 6:79–90.
- Rosenfeldt MT, Ryan KM. 2011. The multiple roles of autophagy in cancer. Carcinogenesis.
- Rowland AA, Voeltz GK. 2012. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat Rev Mol Cell Biol 13:607–625.
- Rubinsztein DC, Shpilka T, Elazar Z. 2012. Mechanisms of autophagosome biogenesis. Curr Biol 22:R29–R34.
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. 2013. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15:741–750.
- Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y. 2009. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 14:525–538.
- Simonsen A, Tooze SA. 2009. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 186:773–782.
- Strømhaug PE, Berg TO, Fengsrud M, Seglen PO. 1998. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J 335(Pt 2):217–224.
- Takahashi Y, Meyerkord CL, Wang HG. 2009. Bif-1/endophilin B1: A candidate for crescent driving force in autophagy. Cell Death Differ 16:947–955.
- Tanida I. 2011. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 14:2201–2214.
- Tanida I, Ueno T, Kominami E. 2008. LC3 and autophagy. Meth Mol Biol 445:77–88.
- Tooze SA, Yoshimori T. 2010. The origin of the autophagosomal membrane. Nat Cell Biol 12:831–835.
- Walker S, Chandra P, Manifava M, Axe E, Ktistakis NT. 2008. Making autophagosomes: Localized synthesis of phosphatidylinositol 3-phosphate holds the clue. Autophagy 4:1093–1096.
- Wang C, Liu Z, Huang X. 2012. Rab32 is important for autophagy and lipid storage in Drosophila. PLoS ONE 7:e32086.
- Wong PM, Puente C, Ganley IG, Jiang X. 2013. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 9:124–137.
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. 2012. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198:219–233.
- Yang Z, Klionsky DJ. 2010a. Eaten alive: A history of macroautophagy. Nat Cell Biol 12:814–822.
- Yang Z, Klionsky DJ. 2010b. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol 22:124–131.
- Yen WL, Legakis JE, Nair U, Klionsky DJ. 2007. Atg27 is required for autophagy-dependent cycling of Atg9. Mol Biol Cell 18:581–593.
- Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 2009. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5:1180–1185.
- Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, et al. 2006. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119:3888–3900.
- Yuan HX, Xiong Y, Guan KL. 2013. Nutrient sensing, metabolism, and cell growth control. Mol Cell 49:379–387.
- Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35.