
Abstract
Sec- and Tat-mediated bacterial lipid modification of proteins are important posttranslational processes owing to their vital roles in cellular functions, membrane targeting and biotechnological applications like ELISA, biosensor, adjuvant-free vaccines, liposomal drug delivery etc. However a better understanding of the tight coupling of secretory and lipid modification machineries and the processes associated will help unravel this essential biological event and utilize it for engineering applications. Further, there is a need for a systematic and convincing investigation into membrane targeting, solubilization and ease-of-purification of engineered lipoproteins to facilitate scientists in readily applying this new protein engineering tool. Therefore, in this study, we have investigated systematically recombinant expression, translocation, solubilization and purification of three White Spot Syndrome Viral (WSSV) proteins, ICP11, VP28 and VP281. Our study shows that the lipid modification and secretion processes are tightly coupled to the extent that mismatch between folding kinetics and signal sequence of target proteins could lead to transcriptional-translational uncoupling or aborted translation. The proteins expressed as lipoproteins through Tat-pathway were targeted to the inner membrane achieving considerable enrichment. These His-tagged proteins were then purified to apparent homogeneity in detergent-free form using single-step Immobilized Metal Affinity Chromatography. This study has interesting findings in lipoprotein biogenesis enhancing the scope of this unique post-translational protein engineering tool for obtaining pure detergent-free, membrane or hydrophobic surface-associating diagnostic targets and vaccine candidates for WSSV.
Introduction
Membrane-associated bacterial lipid-modification is an important posttranslational event owing to its essential roles in cellular functions like adhesion, transport, signal transduction and maintenance of transmembrane potential. Investigations into bacterial lipoproteins and their biogenesis are gaining significance since these play crucial roles in viability, survival and pathogenesis including the ability to trigger innate and adaptive immunity, and the possibilities of useful applications including next generation antibacterial (Babu et al., Citation2006; Fjell et al., Citation2011; Kamalakkannan et al., Citation2005; Kovacs-Simon et al., Citation2011; Kurokawa et al., Citation2012; Shruthi et al., Citation2010b; Sutcliffe et al., Citation2012; Thompson et al., Citation2010; van Bloois et al., Citation2011).
The majority of known bacterial lipoproteins characteristically contain fatty acylated amino terminal, either N-acyl S-diacylglyceryl Cysteine as in Gram-negatives or S-diacylglyceryl Cysteine (Braun & Wu, Citation1994) as in some Gram-positives (Sankaran et al., Citation1995). Recently peptidyl, N-acetyl and lyso forms have also been reported in monoderms (Nakayama et al., Citation2012). This modification consists of three sequential enzymatic reactions (Sankaran & Wu, Citation1994) resulting in the covalent attachment of lipids to the invariant amino terminal Cys of such proteins. Extensive bioinformatic analysis and experimental evidence on bacterial lipid-modification have revealed the requirement of the consensus sequence [LVI][ASTVI][GAS]C, referred to as “lipobox” at the C-terminal of its signal sequence (Babu et al., Citation2006). Partial lipid modification by the first enzyme in the pathway, Lipoprotein diacylglyceryl transferase (Lgt), has been demonstrated in vitro employing a peptide synthesized de novo using the predictive rules (Selvan & Sankaran, Citation2006). Apart from the signal sequence, several other factors and signals essential for the modification and localization have been described (Okuda & Tokuda, Citation2011; Shruthi et al., Citation2010a).
Lipid-phase integration of proteins and export through the cytoplasmic membrane in bacteria occur by the co-translational Signal Recognition Particle (SRP) pathway (Powers & Walter, Citation1997; Saraogi & Shan, Citation2011; Shan & Walter, Citation2005) or posttranslational Sec pathway (Danese & Silhavy, Citation1998). The recently described Twin Arginine Translocase (Tat) pathway (Berks et al., Citation2000; Fröbel et al., Citation2012; Palmer & Berks, Citation2003, Citation2012) is being investigated for its role and mechanism in transporting fast-folding or folded proteins such as cofactor-containing proteins (including lipoproteins) across the cytoplasmic membrane of bacteria; their signal peptide contain an almost invariant twin-arginine motif, S/T-R-R-F-L, between “n” (+ charged N-terminal) and “h” (hydrophobic) regions (Berks, Citation1996).
Bifurcation in the protein secretion pathway (Sec or Tat) depends on the folding kinetics (Pradel et al., Citation2003; Strauch & Georgiou, Citation2007; Whitaker et al., Citation2012). Extensive bioinformatic analysis reported by us provided predictive rules and a tool for identifying Tat-dependent lipoproteins. Our analysis also showed that expression of Tat-dependent lipoproteins is a niche-based adaptation consisting of mostly cofactor-containing or nutrient-binding fast folding proteins (Shruthi et al., Citation2010b). Engineering of lipoproteins like Green Fluorescence Protein (fusion of Tat signal peptide containing Lipobox sequence) indicated the importance of Tat-pathway in the translocation of such fast-folding lipoproteins and indicated that lipid-modification was tightly coupled to translocation (Shruthi et al., Citation2010a).
The current understanding of mechanisms underlying lipid modification in bacteria like E. coli has led to the development of the first posttranslational protein engineering of non-lipoproteins (Kamalakkannan et al., Citation2004). This is enabling man-made applications like the generation of efficacious adjuvant-free vaccine candidates for filariasis (Sharmila et al., Citation2013), membrane-targeting of soluble proteins (Shruthi et al., Citation2010a) and superior diagnostics based on enhanced binding efficiency of lipid-modified protein/peptide ligands to polystyrene surfaces (unpublished work of Shruthi H and Sankaran K). However, there are critical missing links in our understanding as revealed in this study. Therefore systematic study on engineering two envelope proteins (VP28, VP281) and one non-structural histone-binding protein (ICP11) of White Spot Syndrome Virus (which causes mass mortality in shrimps) (Leu et al., Citation2009; Lundin, Citation1996), for bacterial lipid modification has unearthed important and interesting facts about bacterial lipoprotein biosynthesis. This has enabled us to properly design this posttranslational engineering.
Methods
All chemicals were procured either from Sigma (St. Louis, MO), USB Corporation (Cleveland, OH) or Merck (Worli, Mumbai, India). Restriction enzymes and ligase were obtained from New England Biolabs, Inc. (Beverly, MA). The primers used for cloning were synthesized at Eurofins (Bangalore, India). The Genomic and Plasmid DNA purification kits were purchased from Qiagen (Hilden, Germany) and protein purification matrix was acquired from Macherey-Nagel (Duren, Germany) Protein and DNA markers were procured from Fermentas Life Sciences, Germany. E. coli strain GJ1158 was purchased from Bangalore Genei (Bangalore, India), E. coli DH5α and E. coli BL21(DE3) were obtained from Invitrogen (Carlsbad, CA). Nitro cellulose membrane, Hybond-C extra (0.45 micron), was obtained from Amersham Biosciences UK Limited. All the buffers and media were prepared using sterilized double distilled water. Monoclonal anti-his antibody was obtained from GE Health Care Life Sciences (Wipro GE Healthcare Pvt Ltd, Chennai, India). Antiserum against ICP11, VP28 and VP281 were raised in BALB/c mice in-house facility of our laboratory (IAEC No. CBT/AU/IAEC/29/2011) and the IgG was purified using an affinity chromatography column obtained from Merck (Worli, Mumbai, India).
Recombinant constructs for the expression of native and lipid-modified proteins
The total genomic DNA was isolated from experimentally infected White Spot Syndrome Virus P. monodon shrimp using QIAamp DNA Mini Kit (Qiagen, Valencia, CA). The complete open reading frames of two structural (VP28 and VP281) and one non-structural (ICP11) WSSV genes were PCR amplified (Eppendorf Master Cycler). Two sets of PCR amplification primers were used (): (i) FN and RP primers were used for amplification of all the three genes to generate amplicons with NdeI and PstI restriction sites at the N- and C-terminus respectively, and (ii) FB and RP primers were used for amplification of all the three genes to generate amplicons with BamHI and PstI restriction sites at the N- and C-terminus respectively. The amplified products were analyzed by 1% (w/v) agarose gel electrophoresis alongside a 100 bp DNA ladder from Fermentas Life Sciences, Germany.
Table 1. Primers used for cloning of Native, Lipid-modified (Sec & Tat) and Sec mutant recombinant constructs.
The PCR-amplified products with the first set of primers carrying NdeI and PstI sites were treated with respective endonucleases, purified and ligated between the same sites of pRSETB vector (Invitrogen, Carlsbad, CA). The resulting constructs were named as pRSETB-ICP11, pRSETB-VP28 and pRSETB-VP281. Similarly the other set was ligated between the BamHI and PstI sites of modified pRSETB generic vectors namely pG-LM and pG-TL (Kamalakkannan et al., Citation2004; Shruthi et al., Citation2010a) to generate the following lipid-modified constructs with Sec signal peptide, pG-LMICP11, pG-LMVP28 and pG-LMVP281 and with Tat signal peptide, pG-TLICP11, pG-TLVP28 and pG-TLVP281 ().
Figure 1. (A) Schematic representation of the cloning strategy employed to generate constructs, pRSETB-ICP11, pRSETB-VP28, pRSETB-VP281, pG-LMICP11, pG-LMVP28, pG-LMVP281, pG-TLICP11, pG-TLVP28 and pG-TLVP281. (i) Vector map of the prokaryotic expression vector pRSETB in which the genes of native ICP11, VP28 and VP281 were cloned separately between NdeI and PstI sites. (ii) and (iii) Maps of modified prokaryotic expression vector pRSETB showing the fusion of the coding sequence of lipoprotein signal peptide for Sec (pG-LM) and Tat (pG-TL) respectively upstream of target protein’s coding sequence in the MCS between BamHI and PstI sites. (B) The coding sequences of Sec and Tat signal peptide, Lipobox and several residues after invariant lipid-modifiable Cysteine. All the three enlarged versions of the vector maps are enclosed in the Supplementary information (available online only). (C) Agarose gel electrophoresis of the PCR-amplified products of ICP11, VP28 and VP281: (i) PCR-amplified gene products with 5′ NdeI and 3′ PstI sites meant for mature sequence of native proteins, and (ii) PCR-amplified gene products with 5′ BamHI and 3′PstI sites meant for lipid-modification.
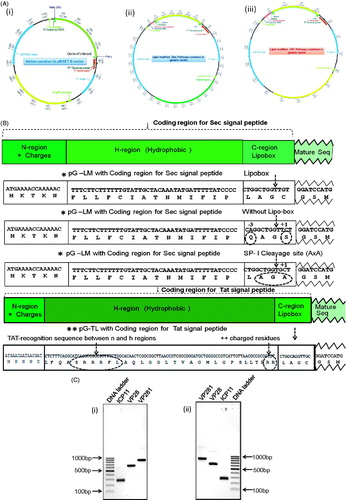
All the above nine recombinant constructs were generated and screened by colony PCR (figure not shown) and further confirmed by DNA sequencing.
Site-directed mutagenesis of pG-LMVP28 and pG-LMVP281
Site-directed mutagenesis in Sec-signal sequence of lipoproteins using mutagenic primers as shown in was performed for VP28 and VP281. The point mutations were introduced in the lipobox at the −3 position: {(Leu (L) → Gln (Q)} and +1 position {Cys (C) → Ser (S)}, changing the two critical residues for lipid modification abolishing the lipobox and ability to undergo lipid modification. In another point mutation, the Cys (C) at +1 position that gets lipid modified was replaced with Ala (A), thus converting lipobox into Signal Peptidase 1 cleavage site (AxA) meant for non-lipoprotein secretory proteins (). After PCR, ligation and transformation for the expression in E. coli GJ1158 and BL21(DE3) were studied after confirming the recombinant construct by DNA sequencing (data not shown).
Expression of native and lipid-modified proteins
All the nine recombinant constructs (3 native and 6 lipid-modified) and corresponding vector (control) were transformed into the E. coli strain, GJ1158 (Genei, Bangalore, India) for the targeted protein expression under the control of salt inducible proU promoter, and transformants were grown at 37 °C in Luria-Bertani Omitted NaCl (LBON) medium containing 100 µg/ml ampicillin (USB corporation, USA.) until the culture reached O.D600nm of 0.6–0.8. The cultures were then induced with 0.3 M NaCl at 37 °C for 4 hours, which resulted in the hyper-expression of the proteins. The whole cells of induced recombinant bacteria were analyzed by 12% SDS-PAGE for the recombinant protein expression and lipid modification.
The site-directed mutagens were transformed into E. coli, GJ1158 and BL21(DE3) for its targeted protein expression and to study the stability of mRNA. The transformants were grown at 37 °C in Luria-Bertani Omitted NaCl (LBON) and Luria-Bertani (LB) medium respectively containing 100 µg/ml ampicillin until the culture reached O.D600 nm of 0.6–0.8. The culture was then induced with 0.3 M NaCl and 1 mM of IPTG respectively at 37 °C for 4 hours. The whole lysate and cellular fractions were analyzed by 12% SDS-PAGE.
Palmitoyl labelling for confirmation of lipid-modification
For palmitate labelling (Shruthi et al., Citation2010a) the native and lipid-modified clones were grown aerobically in 3 ml LBON medium with ampicillin at 37 °C. About 15 minutes prior to O.D600 nm of 0.6 the culture was supplemented with 10 µCi/ml of [9, 10-3H] palmitic acid and then the cultures were induced with 0.3 M NaCl and allowed to grow for another 4 hours in the presence of the label during hyper-expression. Cells harvested by centrifugation at 3000 g for 10 minutes were washed to get rid of the free label and subjected to 12% SDS-PAGE. The separated proteins were transferred to nitrocellulose membrane and exposed to Phosphorimager screen overnight and scanned using Cyclone® Plus Storage Phosphor System, PerkinElmer, to visualize the radiolabeled bands.
RT-PCR to detect gene transcript of engineered VP28 and VP281
The recombinant constructs of VP28 and VP281 were inoculated in LB medium with appropriate antibiotics and incubated in a shaker incubator at 37 °C and 180 rpm until O.D600 reached between 0.8 and 1. The harvested cells were used to extract total RNA using the TRIZOL method (Chirgwin et al., Citation1979; Nicolaides & Stoeckert Jr, Citation1990). An aliquot (5 µl) of RNA was analyzed on a 1.2% (w/v) agarose gel stained with ethidium bromide (25 µg/ml) using 1X TBE (0.04 M Tris borate, 0.001 M EDTA) buffer and examined under a BioRad Gel documentation system. RNA was converted to cDNA using the High Capacity cDNA Reverse Transcription Kits from Applied Biosystems, as per the manufacturer’s recommendations. This cDNA was then used as a template for further amplification by conventional PCR using gene specific primers and the results were analyzed by 1% agarose gel electrophoresis.
Sub-cellular localization by differential detergent solubilization and immunodetection
To identify the sub-cellular localization, cell fractionation was done as described previously (Kamalakkannan et al., Citation2004). Briefly, periplasmic (P), cytoplasmic (C) and total membrane (T) fractions were prepared by lysozyme/EDTA/hypo-osmotic shock and the total membrane proteins were further fractionated to inner membrane (IM) and outer membrane (OM) fractions by ultracentrifugation and further resolved by 12% SDS PAGE to monitor the mobility shift of lipid-modified proteins. The fractionated protein samples from SDS-PAGE gel were then transferred to Nitro-cellulose membrane using Amersham semi-dry transfer apparatus, according to the manufacturer’s recommended procedure. Monoclonal anti-6XHis at a dilution of 1:5000 in phosphate-buffered saline, pH 7.4, followed by alkaline phosphatase conjugated goat anti-mouse IgG secondary antibody (Bangalore Genei) was employed for immunodetection. Washings were performed with phosphate-buffered saline, pH 7.4 and phosphate buffered saline with 0.05% Tween 20, pH 7.4. p-Nitro Blue Tetrazolium Chloride (NBT) and 5-Bromo-4-Chloro-3-Indolyl Phosphate Disodium (BCIP) substrates were used for the visualization of targeted protein bands on the membrane.
Detergent and urea solubilization of engineered lipoproteins
Prior to purification, extraction of bacterial lipid-modified protein from the lipid bilayer was carried out with detergents and urea. In this study the popular mild non-ionic detergent (Octyl-β-D-glucoside) and moderately strong anionic detergent (SLS), which is normally used to solubilize integral inner membrane proteins in active form, were used. Urea was used to probe peripheral membrane association. Total membrane fraction from the induced bacterial cultures (pG-TLICP11, pG-TLVP28 and pG-TLVP281) containing 1.0 mg of total proteins as estimated by the Bradford protein method was taken for solubilization by incubating at 25 °C for 2 hours. Solubilized protein samples were separated by centrifugation at 200 000 g for 1 hour at 4 °C. The soluble and insoluble fractions were analyzed by 12% SDS-PAGE to determine the type of membrane protein, detergent requirement and type of the detergent required for the protein solubilization.
Purification of native and lipid-modified proteins
The hyper-expressed native proteins were purified from the cytosolic fraction and those in the Sec- and Tat-lipid-modified proteins, were purified from the 1% SLS-solubilized fraction. In all the cases, the presence of 6X Histidine tag was utilized for the single-step purification using immobilized metal affinity chromatography (IMAC) as follows. The cell pellets were resuspended in 50 mM sodium phosphate pH 8.0 and 300 mM NaCl and lysed with lysozyme (final concentration 100 µg/mL) and left to stand for 1 hour at 4 °C and sonicated. The membrane fraction was harvested by centrifugation at 150 000 g for 1 hour at 4 °C. The pellet was resuspended in lysis buffer and solubilized with 1% SLS buffer supplemented with 10 mM imidazole, followed by centrifugation for 45 min at 200 000 g at 4 °C. The supernatants containing solubilized membrane proteins were loaded on Tris (carboxymethyl)ethylene diamine (TED) column pre-charged with Ni2+ ion and pre-equilibrated with equilibration buffer supplemented with 10 mM imidazole. The column was then washed with wash buffer containing 20 mM imidazole. The bound-proteins were eluted with purification buffer (50 mM NaH2PO4, 300 mM NaCl and 0.1% SLS pH 8.0) supplemented with 100–250 mM imidazole. The eluted samples were analyzed by 12 % SDS-PAGE. To remove SLS from the purified protein fractions, these were membrane filtered (10 kDa cut-off) with five equal volumes of 50 mM Phosphate buffer pH 8.0. The resulting protein preparations were tested for SLS using Dansyl Cadaverine, capable of detecting 0.001% (w/v) of SLS. Alternately, 10 bed-volumes of washing and elution were done without SLS. The eluted fractions were tested for the detergent by Dansyl Cadaverine assay.
Results
While engineering viral proteins employing previously standardized methods using typical lipoprotein Sec signal sequences, many proteins did not even get expressed, but their native form could be expressed well in the cytosol in soluble form (ICP11) or as inclusion bodies (VP28 and VP281). Our recent study showed that Tat was necessary for both lipid modification and transport of EGFP, a fast-folding protein (Shruthi et al., Citation2010a). Therefore we began a systematic study using three WSSV proteins, ICP11, VP28 and VP281, as a sample in this heterologous lipoprotein engineering. The study probed the requirement of appropriate signal sequence, coupling of the secretory system for not only lipid modification but also expression, and an appropriate engineering system for lipid modification, solubilization and single-step purification.
Tat-dependent expression of lipid-modified WSSV proteins ICP11, VP28 and VP281
The design of the constructs for the expression of native and lipid-modified target proteins is shown in above. In the constructs for native form the ORFs were placed after the ribosome binding site (RBS) in the pRSETB vector. In the constructs for lipid-modification, the coding sequence for Sec or Tat signal peptide containing lipobox was fused with the N-terminal of native ORFs in the former native construct. shows the approximate sizes of the inserts, as expected from the sequence. Once ORFs (249, 615 and 843 bp respectively for ICP11, VP28 and VP281) and the proper fusion were confirmed by DNA sequencing, the roles of the two types of signal sequences in engineered lipoprotein expression were studied in E. coli GJ1158.
As shown in , SDS-PAGE analysis of the whole-cell lysates revealed the presence of ∼11 kDa, ∼28 kDa and ∼37 kDa bands respectively for the native ICP11, VP28 and VP281 and the same fused to Tat signal sequence meant for lipoproteins. No expression could be seen for Sec-VP28 and Sec-VP281, which contained Sec signal sequence for lipoproteins. Lipid-modified ICP11 could be expressed irrespective of the signal sequence type. The authenticity of the expressed proteins was verified by immunoblotting using specific antibodies, as shown in . The lipid modification was indirectly evident from the mild retardation exhibited by the lipo-forms (, compare “Native” and “Tat” lanes). It was further confirmed by metabolic labeling with H3-palmitate; as shown in , proteins engineered for lipid-modification with Sec and Tat signal sequence showed radioactive bands at the expected molecular sizes.
Figure 2. (A) SDS-PAGE profile showing the expression of native and lipid-modified (i) ICP11, and (ii) VP28 and VP281. The corresponding protein migration on the gel is indicated by white arrows. (B) Western blot analysis using specific antibodies showing the expression of the native and lipid-modified ICP11, VP28 and VP281 proteins. (C) Autoradiography of the [9, 10-3H] palmitic acid labelled proteins 1, 2 and 3 correspond to ICP11, VP28 and VP281 respectively, confirming the enzymatic post-translational lipid-modification. Radiolabelling was observed only in the case of Tat-ICP11, Tat-VP28, Tat-VP281 and Sec-ICP11.
![Figure 2. (A) SDS-PAGE profile showing the expression of native and lipid-modified (i) ICP11, and (ii) VP28 and VP281. The corresponding protein migration on the gel is indicated by white arrows. (B) Western blot analysis using specific antibodies showing the expression of the native and lipid-modified ICP11, VP28 and VP281 proteins. (C) Autoradiography of the [9, 10-3H] palmitic acid labelled proteins 1, 2 and 3 correspond to ICP11, VP28 and VP281 respectively, confirming the enzymatic post-translational lipid-modification. Radiolabelling was observed only in the case of Tat-ICP11, Tat-VP28, Tat-VP281 and Sec-ICP11.](/cms/asset/c4932f6b-75df-4394-819a-08f05b9331bb/imbc_a_943819_f0002_b.jpg)
Sec-directed lipid modification did not result in expression of VP28 and VP281
While the expression of ICP11 was observed in both Sec- and Tat-mediated lipid-modification systems, the Sec-mediated lipid-modification of VP28 and VP281 was not observed in both GJ1158 and BL21-(DE3). When the transcription and translation of these recombinants were investigated using qualitative RT-PCR and Western-Immunoblotting respectively, cDNA of expected size could be seen () indicating that the presence of mRNA coding for the above genes. The identity of the transcript was confirmed by sequencing. The lack of expression of Sec-mediated target proteins in the same culture that showed the presence of mRNA was revealed by the Western-immunoblot analysis as shown in , where the bands that corresponded to the lipid-modified VP28 or VP281, could not be seen. To further probe the mechanism underlying this lack of expression, Lipobox sequence was modified by replacing the critical −3 position (L-Q) and +1 position (C-S) residues. This resulted in faint expression for VP28 and moderate expression for VP281, both in the periplasm. Since this suggested that Sec machinery for non-lipoprotein might be involved in processing, signal peptidase I (SP-I), cleavage site was introduced deliberately in place of Lipobox. The expression results were the same. So, lack of expression through Sec-dependent lipoprotein machinery indicated that lipid modification is tightly coupled to translocation. The fact that all the transcripts could be amplified indicated the stability of respective mRNAs ().
Figure 3. (A) Agarose gel electrophoresis confirming the transcription of Sec-VP28 and Sec-VP281 genes (lanes 3 and 5 respectively). In this experiment total RNA was extracted from the Sec-VP28 and Sec-VP281 recombinants in E. coli BL21 (DE3) host and converted into cDNA by RT-PCR, and was further used as a template for PCR using the corresponding primers. (B) Western-Immunoblot analysis of the induced whole cell lysate of E. coli BL21 (DE3) from the same batch of culture used for the above RNA preparation showing that in the case of Sec-mediated lipid-modification of VP28 and VP281 (lanes 2 and 6), the proteins were not even expressed. (C) Agarose gel electrophoresis confirming the transcription of Sec signal peptide without Lipobox and Sec signal peptide with signal peptidase-I cleavage site (AxA) of VP28 and VP281 recombinants constructs. The transcripts of VP28 and VP281 were loaded into lanes 2 & 3 and 6 & 7, lanes 1 and 5 were PCR reaction positive controls of VP28 and VP281 respectively and lane 4 was 100 bp DNA ladder. (D) SDS-PAGE profile showing the expression of VP28 and VP281 with Sec signal peptide without Lipobox and Sec signal peptide with signal peptidase-I cleavage site (AxA). The corresponding protein expression on the gel is indicated by arrows. In the case of VP28 the targeted protein expression was lower the detectable limit by vision. (E) Western-immunoblot analysis confirm the induced, uninduced whole cell lysate of E. coli BL21(DE3) and periplasmic fraction of VP28 and VP281 of targeted proteins. The corresponding protein expression was not detected in uninduced whole cell lysate of E. coli BL21(DE3) and the corresponding proteins were secreted in the periplasmic space.
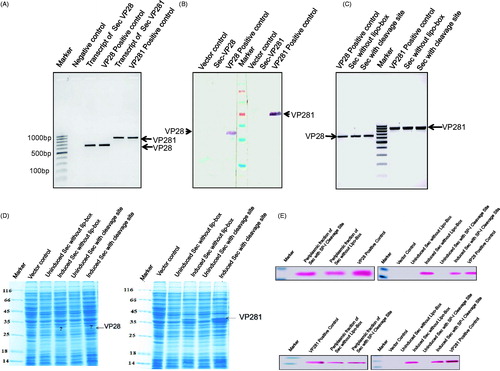
Table 2. Comprehensive analysis of protein expression, its translocation and stability of mRNA.
All the Tat-secreted lipid-modified proteins were localized to Sarkosyl-soluble (inner membrane) fraction
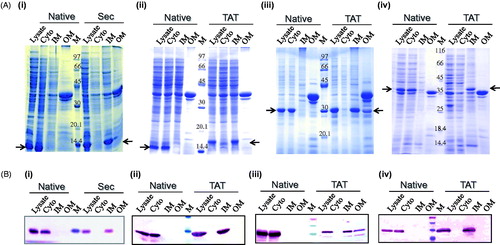
Sub-cellular fractions, cytoplasm, inner membrane and outer membrane, examined by SDS-PAGE and Western-immunoblot analysis showed that the native proteins (expressed from the constructs pRSETB-ICP11, pRSETB-VP28 and pRSETB-VP281) were localized to the cytoplasm. Sec-mediated ICP11 and Tat-mediated ICP11, VP28 and VP281 were found to be localized to the Sarkosyl-soluble fraction, the inner membrane. Tat-mediated VP28 exhibited partial solubilization requiring Sarkosyl-solubilization twice. Protein localization was further ascertained by immunoblotting using monoclonal anti-VP28 antibody and monoclonal anti-His antibody for all other proteins. As can be seen in , the immunoreactive bands corresponded to the sub-cellular fractions deduced from Coomassie blue stained sub-cellular protein profiles ().
Figure 4. (A) SDS-PAGE profile of 100 mg of all fractions showing the expression and sub-cellular localization (see arrows) of (i) Sec-ICP11, (ii) Tat-ICP11, (iii) Tat-VP28, and (iv) Tat-VP281 cloned with (for lipid modification) and without (native) the corresponding signal peptide. All the native proteins were expressed in the cytosol (see second lane in all the pictures). The lipid-modified proteins were localized to the inner membrane (see 8th lane in all the pictures). (B) Western-immunoblot analysis to ascertain the sub-cellular localization of the respective proteins. Monoclonal anti-VP28 antibody was used for probing VP28 proteins and monoclonal anti-his antibody for the rest of the proteins. Cyto, cytoplasm; IM, inner membrane; OM, outer membrane; M, marker.
The lipid-modified viral proteins required moderately strong detergent for solubilization
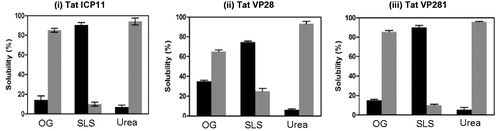
One of the critical aspects of this type of protein engineering by localizing proteins to membrane is about the strength of association and detergent solubility and the possible difficulties in getting rid of the detergent subsequent to purification. This was studied by employing Urea, a non-detergent membrane protein solubilizing agent, Octylglucoside, a mild detergent, and Sarkosyl, a moderately strong detergent. As seen in , in all the cases, only Sarkosyl was able to completely solubilize the engineered lipoproteins, except VP28, which required solubilization twice. Urea could not solubilize at all, as expected for integral proteins. Interestingly, Octylglucoside, which could not significantly solubilize other engineered proteins, could solubilize about 25% of lipid-modified VP28, which actually required Sarkosyl solubilization twice. This signifies the protein-specific contributions in the association of lipid anchor to the membrane.
Figure 5. Bar graph from the triplicate experiment representing the extent of solubilization of lipid-modified (i) Tat-ICP11, (ii) Tat-VP28, and (iii) Tat-VP281 in different solubilizing agents: β-Octyl glucoside (OG), Sodium lauoryl sarcocine (SLS) and Urea. The black bars represent the percentage of soluble fraction and the grey bars represent the percentage of insoluble fraction.
His-tagged engineered lipoproteins could be purified in a single-step using IMAC in detergent-free form
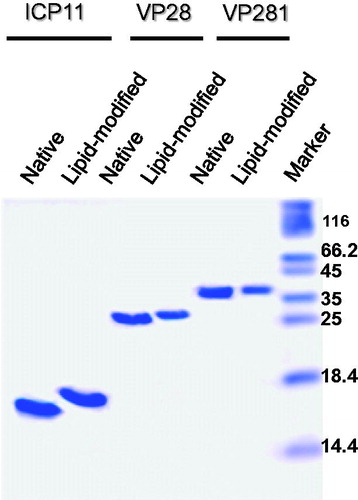
To address the concerns of protein purification of such engineered lipoproteins from detergent solution, all the three recombinant lipoproteins were purified using IMAC exploiting the His-tag at their C-terminal. The concentration of the detergent was also reduced 10 times during elution and further removed by membrane filtration. The same result could be obtained when the column was washed and eluted under SLS-free condition. No visible precipitation was seen and all the protein could be recovered in the supernatant after ultracentrifugation at 200 000 g for 1 hour (data not shown). As can be seen from the purified proteins were apparently homogenous by Commassie blue staining after SDS-PAGE. The tricine gel showed the expected retardation of lipid-modified proteins indicating the preservation of the characteristic of lipoproteins. Chemical analysis for Sarkosyl using Dansyl Cadaverine (Lee et al., Citation1989) showed the amount of SLS that remained is lower that 0.001 % (w/v) either after membrane filtration or after washing and elution without detergent. Therefore it appeared that engineered lipoproteins could be readily purified in a detergent-free form in a single-step, if the washing and elution steps are done devoid of detergent in affinity chromatography, or using steps standardized for native proteins.
Figure 6. Tricine-SDS-PAGE profile showing the migration difference between purified native (lanes 1,3,5) and lipid-modified (lanes 2,4,6) ICP11, VP28 and VP281. Apparently homogenous preparations in the soluble state can be deduced from the analysis of 5 µg (VP28 and VP281) and 10 µg (ICP11) proteins loaded.
Discussion
Posttranslational lipid modification of proteins in bacteria is still not fully understood. Biologically important aspects like the interactions between lipid modification and transport machineries and its significance in the biogenesis remain inadequately probed. Such knowledge is also essential for bio-conjugation of proteins with lipids, which is an emerging protein engineering solution for a variety of applications including targeted drug delivery and biosensor-based diagnostic applications (Sawant & Torchilin, Citation2011). In this context, this unique and ubiquitous lipid modification of proteins in bacteria is utilized as a novel protein engineering tool in our laboratory to develop biotech applications like enhanced efficiency (4–5 orders better than the native protein) of lipid-modified proteins/peptides to bind polystyrene surfaces in ELISA (unpublished work of Shruthi H and Sankaran K), membrane targeting of cytosolic and periplasmic proteins (Kamalakkannan et al., Citation2004; Shruthi et al., Citation2010a) and development of efficacious adjuvant-free vaccine candidate for filariasis (Sharmila et al., Citation2013). In the process, new knowledge has emerged about the signals, machineries and regulatory factors that govern bacterial lipid modification. In spite of this advancement, inadequacies in our understanding of bacterial lipoprotein biogenesis became apparent when we tried to hyper-express lipid-modified WSSV proteins and other viral proteins as targets for diagnostics and therapeutics of viral diseases. Moreover, there is no systematic study on the hyper-expression, solubilization and purification of such engineered lipoproteins addressing major concerns like the ability to obtain detergent-free soluble protein preparations in a simple procedure, preferably by a single-step. We have addressed these in this study and it is hoped that this would open up avenues for the growth of this interesting posttranslational modification and new biotechnology tool.
Important links between protein translocation and lipid-modification machineries have only recently been investigated in vivo to understand bacterial lipoprotein biosynthesis. It was proposed that the translocation-competent and translocase-associated precursor form is a prerequisite for the initiation of lipid-modification in vivo (Shruthi et al., Citation2010a). In this study, for the first time, we have also investigated the roles of Sec- and Tat-lipoprotein signal peptides on the expression and targeting of three proteins, ICP11, VP28 and VP281, in lipid-modified form to various sub-cellular compartments in recombinant bacterial hosts. Taking this as a random set of proteins for heterologous lipid modification, the lessons learned are valuable in the improvement and application of this novel protein engineering.
The expression of all the three proteins in their native forms led to either inclusion bodies (VP28, VP281) or soluble forms (ICP11) localized in the cytosol. With the exception of ICP11, the proteins were expressed only when transported through Tat machinery, but not Sec machinery. Interestingly, though VP28 and VP281 could not be expressed at all with Sec signal, their mRNA could still be isolated. The failure to get the message translated indicated uncoupling of transcription and translation suggesting translational control when appropriate signal is not associated with the right folding kinetics. The lack of right signals in the mRNA, influence of ribosomes on mRNA decay, R-loop formation, endonuclease activity on the nascent RNA, RNA secondary structure formation, and interaction of termination factor Rho and elongation factor NusG are some of the possible uncoupling mechanisms reported in the literature (Deana & Belasco, Citation2005; Gowrishankar & Harinarayanan, Citation2004; Leela et al., Citation2013; Kudla et al., Citation2009; Saxena & Gowrishankar, Citation2011). The mRNA secondary structure affecting the translation initiation could be ruled out because the three recombinant gene constructs had an identical coding region for the signal peptides and were cloned in the same vector in an identical manner. Considering the other possibilities, our hypothesis would be that in a co-translational transport-coupled modification, mismatch of type of signal sequence with folding kinetics (in this case fast-folding Tat substrates, VP28 and VP281, with Sec signal for unfolded/slow-folding lipoproteins) could jam the Sec pathway affecting the essential general transport of proteins across the cytoplasmic membrane. To avoid such mishaps the cells might have evolved this type of translational control to uncouple transcription and translation. Lack of expression in case of VP28 and VP281 when mediated through Sec signal sequence with lipobox, but mild expression of VP28 and moderate expression of VP281 targeted to the periplasm when the lipobox was modified or replaced with SP-I cleavage site suggested that lipid modification is more tightly coupled to translation, translocation and processing of signal peptide than the Sec machinery for non-lipoproteins. Whether it is aborted translocation or uncoupling of transcription and translocation is not clear without radioactive pulse-chase experimental results. The ability of SecA to bind to Sec-signal peptides and guide co-translational transport through Sec (Karamyshev & Johnson, Citation2005), transcriptional regulation of SecA expression through transcription-translation uncoupling by Sec M (Yap & Bernstein, Citation2011) (and the structural differences between the Sec and Tat signal peptides (Berks et al., Citation2003) are relevant to the hypothesis. Further study to understand this aspect will be rewarding in terms of regulation of protein expression and bacterial homeostasis.
In the case of ICP11, a small unstructured protein of 11 kDa, Sec- as well as Tat-guided expression and lipid modification was possible, even though two different signals were present for the protein with the same folding kinetics. The exact reason cannot be elucidated from the present data. It may be worthwhile to consider the fact that in Sec-mediated lipoprotein biosynthesis small proteins like murein lipoproteins do not require SecB (Sugai & Wu, Citation1992) and kinetic partitioning between folding and association (Kim & Kendall, Citation2000) determines SecB binding. Similarly it is plausible that smaller unstructured protein-like ICP11 could be an exception to the rule associating secretion machinery with folding kinetics (it may be relevant to recall that unlike native VP28 and VP281, ICP11 could be expressed in soluble form in the cytosol) as mentioned above. However, more insightful work is needed to provide the convincing proof.
In our previous study we had hypothesized that the Tat-mediated pathway helped in the translocation of lipid-modified fast-folding proteins like EGFP to the outer membrane (Shruthi et al., Citation2010a). In this study we found that all the three Tat-mediated lipid-modified proteins were targeted to the inner membrane and none to the outer membrane, as evidenced by Sarkosyl solubilization. Whereas ICP11 is an unstructured soluble cytosolic protein, VP28 is a beta-barrel envelope protein with its N-terminal helix (Tang et al., Citation2007), which forms inclusion bodies during hyper-expression. The structure of VP281 is not known. It has been reported that +2, +3 positions after the N-terminal lipid-modifiable Cys have targeting roles (Terada et al., Citation2001). Gln at the +2 position is expected to target proteins to the outer membrane. The targets in this study had no such features; the +2 and +3 positions were Ser as in the case of beta barrel protein, EGFP, whose lipo-form was targeted to the outer membrane (Shruthi et al., Citation2010a). Then why all the three proteins were targeted to the inner membrane, including the beta barrel protein, VP28, is intriguing. It has also been reported that many Tat-proteins are periplasmic and not targeted to the outer membrane. Taken together, this study points out that additional unknown structural features determine the localization of native or engineered Tat-lipoproteins. Further detailed investigation to identify the structural elements responsible for outer membrane targeting is underway.
It is well-known that SLS has the capability to selectively solubilize inner membrane proteins but not the outer membrane proteins. This was true in the case of lipid-modified proteins too, even though these are associated with the membrane through the three fatty acyl chains rather than transmembrane peptide(s). Even more interestingly, when engineered, lipid-modified EGFP got targeted to the outer surface of the outer membrane of E. coli and could be solubilized only with SDS, a strong denaturing detergent. This indicated that detergent solubilization of membrane proteins is decided by the composition of the milieu they are in. The partial solubilization of lipid-modified VP28 in SLS and increased solubility in the mild detergent Octyl glucoside, which was not effective in the case of other proteins, can be attributed to the protein’s structure and anomalous membrane association.
A major challenge in working with membrane proteins is their preparation without losing activity or containing detergents. Since bacterial lipid-modification facilitates engineered hydrophilic proteins to be anchored to the membrane through its N-terminal acyl chains, it is not expected to pose the same problems as the transmembrane peptides do. After all, lipoproteins are nature’s adaptation for hydrophilic proteins, which are quite soluble and can be maintained in detergent-free conditions. In essence, our study could achieve single-step purification of His-tagged lipoproteins using metal affinity chromatography after partially enriching them by targeting the inner membrane. The fact that detergent could be diluted out or removed during affinity purification (or other chromatography steps based on the native protein structure and function) or completely removed by filtration showed that detergent-free pure lipoprotein preparation could be readily and effortlessly obtained, as in this case to get diagnostic targets and vaccine candidates with biological membrane or hydrophobic surface-binding properties.
Conclusions
The conclusion of our study on recombinant lipid modification of three WSSV proteins (ICP11, VP28 and VP281) reveals the necessity of using proper (Sec or Tat) signal peptide for protein lipid modification. When there is a mismatch between folding kinetics and the signal peptide type plausibly transcription translation uncoupling results with no expression, an observation made for the first time in lipoprotein biogenesis. The engineered lipoproteins targeted to the inner membrane were solubilized in SLS and were readily purified to apparent homogeneity in a single step by exploiting the C-terminal His-tag and IMAC systems, and rendering them detergent-free and soluble.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
We gratefully acknowledge the Department of Biotechnology, Government of India, for supporting the work through InNoVacc and the Centre of Excellence Programme (Grant number: BT/AAQ/Indo-Norway/183196/2007/ Date 15.12.2008). The fellowship to Mr S. Kumar from InNoVacc project is gratefully acknowledged.
Supplementary Material Available Online
Supplementary Figures 1, 2 and 3
Supplementary Material
Download PDF (1.7 MB)Acknowledgements
We thank (the late) Dr V. Murugan, Centre for Biotechnology, Anna University for his initial contributions and we dedicate the work to his memory.
References
- Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, Sankaran K. 2006. A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188:2761–2773
- Berks BC. 1996. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol 22:393–404
- Berks BC, Palmer T, Sargent F. 2003. The Tat protein translocation pathway and its role in microbial physiology. Adv Microbial Physiol 47:187–254
- Berks BC, Sargent F, Palmer T. 2000. The Tat protein export pathway. Mol Microbiol 35:260–274
- Braun V, Wu H. 1994. Lipoproteins, structure, function, biosynthesis and model for protein export. New Comprehen Biochem 27:319–341
- Chirgwin JM, Przybyla AE, Macdonald RJ, Rutter WJ. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18:5294–5299
- Danese PN, Silhavy TJ. 1998. Targeting and assembly of periplasmic and outer-membrane proteins in Escherichia coli. Ann Rev Genet 32:59–94
- Deana A, Belasco JG. 2005. Lost in translation: The influence of ribosomes on bacterial mRNA decay. Genes Devel 19:2526–2533
- Fjell CD, Hiss JA, Hancock RE, Schneider G. 2011. Designing antimicrobial peptides: Form follows function. Nature Rev Drug Discovery 11:37–51
- Fröbel J, Rose P, Müller M. 2012. Twin-arginine-dependent translocation of folded proteins. Philosoph Trans Royal Society B: Biol Sci 367:1029–1046
- Gowrishankar J, Harinarayanan R. 2004. Why is transcription coupled to translation in bacteria? Mol Microbiol 54:598–603
- Kamalakkannan S, Murugan V, Jagannadham M, Nagaraj R, Sankaran K. 2004. Bacterial lipid modification of proteins for novel protein engineering applications. Protein Eng Design Selection 17:721–729
- Kamalakkannan S, Murugan V, Sankaran K. 2005. Bacterial lipid modification of proteins and its potential applications. BIOforum Europe 3:44–47
- Karamyshev AL, Johnson AE. 2005. Selective SecA association with signal sequences in ribosome-bound nascent chains: A potential role for SecA in ribosome targeting to the bacterial membrane. J Biolog Chem 280:37930–37940
- Kim J, Kendall DA. 2000. Sec-dependent protein export and the involvement of the molecular chaperone SecB. Cell Stress Chaperones 5:267–275
- Kovacs-Simon A, Titball R, Michell SL. 2011. Lipoproteins of bacterial pathogens. Infection Immunity 79:548–561
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324:255–258
- Kurokawa K, Ryu KH, Ichikawa R, Masuda A, Kim MS, Lee H, et al. 2012. Novel bacterial lipoprotein structures conserved in low-GC content Gram-positive bacteria are recognized by Toll-like receptor 2. J Biolog Chem 287:13170–13181
- Lee YM, Nakamura H, Nakajima T. 1989. Fluorometric determination of carboxylic performance liquid chromatography after with onodansyl cadaverine. Analyt Sci 5:681–685
- Leela JK, Syeda AH, Anupama K, Gowrishankar J. 2013. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc Natl Acad Sci 110: 258–263
- Leu JH, Yang F, Zhang X, Xu X, Kou GH, Lo CF. 2009. Whispovirus. In Van Etten JL, ed. Lesser Known Large dsDNA Viruses. Berlin Heidelberg: Springer, 197–227
- Lundin G. 1996. Global attempts to address shrimp disease. Marine/Environmental Paper No. 4. Land, Water and Natural Habitats Division, Environment Department, The World Bank, Rome, 45
- Nakayama H, Kurokawa K, Lee BL. 2012. Lipoproteins in bacteria: Structures and biosynthetic pathways. FEBS J 279:4247–4268
- Nicolaides N, Stoeckert JRC. 1990. A simple, efficient method for the separate isolation of RNA and DNA from the same cells. Biotechniques 8:154–156
- Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Ann Rev Microbiol 65:239–259
- Palmer T, Berks BC. 2003. The Tat protein export pathway. Protein secretion pathways in bacteria. Springer Netherlands, 51–64
- Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nature Rev Microbiol 10:483–496
- Powers T, Walter P. 1997. Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. EMBO J 16:4880–4886
- Pradel N, Santini CL, Ye CY, Fevat L, Gérard F, Alami, M, Wu LF. 2003. Influence of tat mutations on the ribose-binding protein translocation in Escherichia coli. Biochem Biophys Res Commun 306:786–791
- Sankaran K, Gupta SD, Wu HC. 1995. Modification of bacterial lipoproteins. Meth Enzymol 250:683–697
- Sankaran K, Wu HC. 1994. Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. J Biolog Chem 269:19701–19706
- Saraogi I, Shan SO. 2011. Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic 12:535–542
- Sawant RR, Torchilin VP. 2011. Design and synthesis of novel functional lipid-based bioconjugates for drug delivery and other applications. In Mark SS, ed. Methods in Molecular Biology in Bioconjugation Protocols, Vol. 751. Heidelberg, Germany: Springer, 357–378
- Saxena S, Gowrishankar J. 2011. Modulation of Rho-dependent transcription termination in Escherichia coli by the H-NS family of proteins. J Bacteriol 193:3832–3841
- Selvan AT, Sankaran K. 2006. Bacterial lipid modification in vitro: Synthetic peptide substrate for phosphatidylglycerol-prolipoprotein diacylglyceryl transferase. Indian J Biotechnol 5:327–331
- Shan SO, Walter P. 2005. Co-translational protein targeting by the signal recognition particle. FEBS Lett 579:921–926
- Sharmila S, Christiana I, Kiran P, Reddy MV, Sankaran K, Kaliraj P. 2013. Bacterial lipid modification enhances immunoprophylaxis of filarial Abundant Larval Transcript-2 protein in Mastomys model. Parasite Immunol 35:201–213
- Shruthi H, Anand P, Murugan V, Sankaran, K. 2010a. Twin arginine translocase pathway and fast-folding lipoprotein biosynthesis in E. coli: Interesting implications and applications. Mol BioSyst 6:999–1007
- Shruthi H, Babu MM, Sankaran K. 2010b. TAT-pathway-dependent lipoproteins as a niche-based adaptation in prokaryotes. J Mol Evol 70:359–370
- Strauch EM, Georgiou G. 2007. A bacterial two-hybrid system based on the twin‐arginine transporter pathway of E. coli. Protein Sci 16:1001–1008
- Sugai M, Wu H. 1992. Export of the outer membrane lipoprotein is defective in secD, secE, and secF mutants of Escherichia coli. J Bacteriol 174:2511–2516
- Sutcliffe IC, Harrington DJ, Hutchings MI. 2012. A phylum level analysis reveals lipoprotein biosynthesis to be a fundamental property of bacteria. Protein Cell 3:163–170
- Tang X, Wu J, Sivaraman J, Hew CL. 2007. Crystal structures of major envelope proteins VP26 and VP28 from white spot syndrome virus shed light on their evolutionary relationship. J Virol 81:6709–6717
- Terada M, Kuroda T, Matsuyama SI, Tokuda H. 2001. Lipoprotein sorting signals evaluated as the LolA-dependent release of lipoproteins from the cytoplasmic membrane of Escherichia coli. J Biolog Chem 276:47690–47694
- Thompson BJ, Widdick DA, Hicks MG, Chandra G, Sutcliffe IC, Palmer T, Hutchings MI. 2010. Investigating lipoprotein biogenesis and function in the model Gram-positive bacterium Streptomyces coelicolor. Mol Microbiol 77:943–957
- Van Bloois E, Winter RT, Kolmar H, Fraaije MW. 2011. Decorating microbes: Surface display of proteins on Escherichia coli. Trends Biotechnol 29:79–86
- Whitaker N, Bageshwar UK, Musser SM. 2012. Kinetics of precursor interactions with the bacterial Tat translocase detected by real-time FRET. J Biolog Chem 287:11252–11260
- Yap MN, Bernstein HD. 2011. The translational regulatory function of SecM requires the precise timing of membrane targeting. Mol Microbiol 81:540–553