
Abstract
Mutations in human LMBRD1 and ABCD4 prevent lysosomal export of vitamin B12 to the cytoplasm, impairing the vitamin B12-dependent enzymes methionine synthase and methylmalonyl-CoA mutase. The gene products of LMBRD1 and ABCD4 are implicated in vitamin B12 transport at the lysosomal membrane and are proposed to act in complex. To address the mechanism for lysosomal vitamin B12 transport, we report the novel recombinant production of LMBD1 and ABCD4 for detailed biophysical analyses. Using blue native PAGE, chemical crosslinking, and size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS), we show that both detergent-solubilized LMBD1 and detergent-solubilized ABCD4 form homodimers. To examine the functional binding properties of these proteins, label-free surface plasmon resonance (SPR) provides direct in vitro evidence that: (i) LMBD1 and ABCD4 interact with low nanomolar affinity; and (ii) the cytoplasmic vitamin B12-processing protein MMACHC also interacts with LMBD1 and ABCD4 with low nanomolar affinity. Accordingly, we propose a model whereby membrane-bound LMBD1 and ABCD4 facilitate the vectorial delivery of lysosomal vitamin B12 to cytoplasmic MMACHC, thus preventing cofactor dilution to the cytoplasmic milieu and protecting against inactivating side reactions.
Introduction
Dubbed nature’s most beautiful cofactor (Stubbe, Citation1994), cobalamin (Cbl or vitamin B12) is the largest of all vitamins and the most structurally complex (Hodgkin et al., Citation1956). It is composed of a central cobalt atom that is coordinated by a tetrapyrrole framework termed the corrin ring and by two axial ligands. The lower axial ligand is in a “base-on” state when the pendant dimethylbenzimidazole (DMB) base, connected to the corrin ring, coordinates cobalt. Protonation of DMB or histidine substitution results in the “base-off” or “base-off/His-on” forms of Cbl, respectively. Cbl diversity is provided by its upper axial ligand that covalently attaches to the central cobalt. Methyl, 5′-deoxyadenosyl, hydroxyl, or cyano groups can serve as upper axial ligand, giving rise to several forms of Cbl: methylcobalamin (MeCbl), 5′-adenosylcobalamin (AdoCbl), hydroxocobalamin (OHCbl), and cyanocobalamin (CNCbl). Methylmalonyl-CoA mutase (MCM; EC 5.4.99.2) and methionine synthase (MS; EC 2.1.1.13) use Cbl as cofactor to catalyze chemically unique reactions. Methionine is generated from homocysteine by cytoplasmic MS and MeCbl. AdoCbl is used by mitochondrial MCM for the isomerization of methylmalonyl-CoA to succinyl-CoA for entry into the tricarboxylic acid (TCA) cycle.
Nine inherited defects of intracellular Cbl metabolism, designated cblA to cblG, cblJ and mut, have been discovered through somatic complementation assays (Coelho et al., Citation2012; Froese & Gravel, Citation2010). Defects in this pathway result in MCM and MS substrate accumulation; the resultant methylmalonic aciduria and/or homocystinuria can give rise to severe hematological and neurological conditions. Patients within the cblF (Rosenblatt et al., Citation1985; Watkins & Rosenblatt, Citation1986) or cblJ (Coelho et al., Citation2012; Kim et al., Citation2012) groups have similar clinical phenotypes: hypotonia, lethargy, poor feeding, bone marrow suppression, macrocytic anemia and heart defects. Cultured fibroblasts from cblF patients accumulate nonprotein-bound Cbl in lysosomes (Rosenblatt et al., Citation1985; Vassiliadis et al., Citation1991). The accumulation of unprocessed, nonprotein-bound Cbl within the cell has also been reported for cblJ patients (Coelho et al., Citation2012; Kim et al., Citation2012) who mimic the cblF phenotype (Rosenblatt et al., Citation1985). Mutations in LMBRD1 (Rutsch et al., Citation2009) and ABCD4 (Coelho et al., Citation2012) were recently identified as the cause of the cblF and cblJ defects, respectively. Transfection of LMBRD1 into cblF fibroblasts or ABCD4 into cblJ fibroblasts rescues AdoCbl and MeCbl synthesis and restores MCM and MS function, implicating these gene products in the lysosomal export of Cbl (Coelho et al., Citation2012; Rutsch et al., Citation2009).
LMBRD1 encodes LMBD1, a 61.4 kDa membrane protein of 540 residues and six predicted lysosomal N-glycosylation sites (Rutsch et al., Citation2009). LMBD1 localizes primarily to the lysosome (Chapel et al., Citation2013; Rutsch et al., Citation2009) but also appears in the plasma membrane (Tseng et al., Citation2013) and perinuclear region (Rutsch et al., Citation2009). Based on topology modeling programs, predicted glycosylation pattern and freeze-fracture replica immunolabeling experiments (Rutsch et al., Citation2009), LMBD1 spans the lysosomal membrane via nine transmembrane (TM) helices with a lysosomal N-terminus and cytoplasmic C-terminus. LMBD1 demonstrates homology to the lipocalin-1 interacting membrane receptor (LIMR) involved in the internalization of lipocalins that bind small hydrophobic molecules like steroids and lipids (Rutsch et al., Citation2009, Citation2011).
ABCD4 encodes ABCD4, a non-glycosylated protein of 606 residues with molecular mass of 68.6 kDa (Holzinger et al., Citation1997; Kashiwayama et al., Citation2009). While ABCD4 was originally identified as peroxisomal (Shani et al., Citation1997), recent studies have suggested its localization to the ER (Kashiwayama et al., Citation2009) or within the lysosome of human beings (Coelho et al., Citation2012) and rats (Chapel et al., Citation2013). ABCD4 consists of two domains: an N-terminal TM domain and a C-terminal nucleotide-binding domain (NBD) (Coelho et al., Citation2012). The TM domain has six predicted TM helices (Coelho et al., Citation2012; Kashiwayama et al., Citation2009) whereas the NBD contains highly conserved motifs involved in ATP and Mg2+ binding, including Walker A and B motifs plus a modified ATP-binding cassette (ABC) signature motif (Coelho et al., Citation2012). Based on bioinformatics predictions and loop-directed glycosylation experiments, the topology of ABCD4 has both the N- and C-termini exposed to the cytoplasm (Coelho et al., Citation2012; Kashiwayama et al., Citation2009). ABCD4 is a member of the D subfamily of ABC half transporters that dimerize to form active, full transporters (Hillebrand et al., Citation2007; Liu et al., Citation1999). ABCD4 has significant amino acid identity (25–27%) with these other members, but lacks a hydrophilic N-terminal tail that mediates localization to the peroxisome (Kashiwayama et al., Citation2009).
The functional interplay between LMBD1 and ABCD4 was originally proposed by a fluorescence microscopy study that demonstrated colocalization of both proteins to the lysosomal membrane (Coelho et al., Citation2012). Further supported by partial functional rescue by transfection of ABCD4 cDNA into cultured cblF fibroblasts (Coelho et al., Citation2012), it is suggested that ABCD4 and LMBD1 form a complex that is critical to the export of lysosomal Cbl to the cytoplasm. These lysosomal membrane proteins are also proposed to mediate the directional transfer of Cbl to the downstream cytoplasmic Cbl-processing protein MMACHC (Froese & Gravel, Citation2010; Rutsch et al., Citation2011).
This study provides detailed characterizations of the protein interactions involved in Cbl egress from the lysosome. To our knowledge, this is the first description of recombinant LMBD1 and ABCD4 production. Exploiting blue native PAGE (BN-PAGE), chemical crosslinking, and size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS), we determined that detergent-solubilized LMBD1 and ABCD4 formed homodimers in solution. Adopting label-free, real-time surface plasmon resonance (SPR), LMBD1 and ABCD4 interacted with low nanomolar affinity. As predicted by phage display, SPR also confirmed that MMACHC can interact with both LMBD1 and ABCD4 with low nanomolar affinities. Our results are consistent with a model that implicates LMBD1 and ABCD4 in the vectorial delivery of lysosomal Cbl to cytoplasmic MMACHC. Our direct evidence for protein interactions involved in egress of lysosomal Cbl advances basic understanding of early intracellular Cbl metabolism that, to date, has largely been limited to studies in cultured cells.
Methods
Protein purification
cDNA corresponding to LMBRD1 or ABCD4 were cloned into the pFB-CT10HF-LIC transfer vector that encodes downstream sequences in the following order: a TEV cleavage site (ENLYFQS); a decahistidine tag (His10); and a FLAG (DYKDDDDK) tag. Baculoviruses were constructed according to the Bac-to-Bac system (Life Technologies). Spodoptera frugiperda (Sf9) insect cells were grown in shaker flasks in I-MAX (Wisent) or Sf-900 II SFM media (Life Technologies) at 27 °C and infected with amplified recombinant baculovirus (MOI of 0.25–0.5) for 72 h. Following infection, cells were collected by centrifugation (1000 g for 10 min), washed in PBS, flash frozen in liquid nitrogen, and stored at −80 °C.
Thawed, infected cells were resuspended (25 ml/L of cells) in cold lysis buffer (50 mM HEPES pH 7.5 plus a Complete EDTA-free protease inhibitor cocktail tablet [Roche]) and lysed by three passes through an EmulsiFlex-C5 homogenizer (Avestin) at 15 000 psi. Supernatants collected following centrifugation (10 000 g at 4 °C for 10 min) were supplemented with NaCl to a final concentration of 1 M. Membranes were recovered by ultracentrifugation (100 000 g at 4 °C for 75 min) and resuspended in lysis buffer plus 1 M NaCl using a Dounce homogenizer. After an additional round of ultracentrifugation (100 000 g at 4 °C for 75 min), membranes expressing LMBD1 were resuspended in extraction buffer [50 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol, 1% (w/v) n-dodecyl-β-D-maltoside (DDM) and 0.1% (w/v) cholesteryl hemisuccinate (CHS)]. Membranes expressing ABCD4 were treated with extraction buffer plus 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) and 1.0 mM MgCl2 for 16 h at 4 °C by slow mixing on a rotating mixer.
Detergent-extracted membranes collected after ultracentrifugation (100 000 g at 4 °C, 45 min) were supplemented with 10 mM imidazole. Ni-NTA Superflow (Qiagen), pre-equilibrated in purification buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol, 0.03% DDM, 0.003% CHS, plus 0.5 mM TCEP and 1 mM MgCl2 for ABCD4 preparations) with 10 mM imidazole, was added to detergent-extracted membranes, followed by gentle mixing for 1 h at 4 °C. Resin was captured through a gravity column and washed with 15 column volumes (CV) of purification buffer plus 20 mM imidazole, followed by stringent washing using 15 CV of purification buffer supplemented with 40 mM imidazole. Protein was eluted by addition of purification buffer containing 300 mM imidazole and exchanged into gel filtration buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 0.02% DDM, 0.002% CHS, plus 0.5 mM TCEP and 1.0 mM MgCl2 for ABCD4), using a PD Minitrap G-25 desalting column (GE Healthcare Life Sciences). TEV protease was added to each sample in a 1:2 mass ratio of TEV:protein, and Peptide-N-Glycosidase F (PNGase F) was concurrently added to LMBD1 samples in a 1:30 mass ratio of PNGaseF:LMBD1 and incubated for 72 h at 4 °C.
Cleaved LMBD1 and ABCD4 were separated from His6-tagged TEV protease and PNGase F by incubation for 1 h with Ni-NTA Superflow resin at 4 °C. Resin was collected in a gravity column and flow through was pooled and concentrated to ∼1 mg/ml using a centrifugal filter (Millipore; Amicon 100 kDa MWCO). MMACHC and MMADHC were cloned, expressed, and purified as previously described (Deme et al., Citation2012; Plesa et al., Citation2011). Protein concentrations were derived from A280 measurements using the theoretical extinction coefficients and molecular masses for LMBD1 (106 230 M−1 cm−1; 62.2 kDa), ABCD4 (92 250 M−1 cm−1; 69.4 kDa), MMACHC (47 900 M−1 cm−1; 31.9 kDa), and MMADHC (32 430 M−1 cm−1; 31.9 kDa).
Purity of LMBD1 and ABCD4 was assessed by SDS-PAGE using 8% polyacrylamide gels followed by silver staining. Apparent protein molecular masses derived from SDS-PAGE were estimated using molecular mass standards. Monodispersity of LMBD1 and ABCD4 protein-detergent complexes (PDCs) were assessed using analytical size exclusion chromatography (aSEC). Briefly, samples (50 μg) were applied onto a Superdex 200 10/300 GL column (GE Healthcare Life Sciences) equilibrated in gel filtration buffer and apparent molecular masses were derived from soluble protein standards (Bio-Rad). Purified LMBD1 and ABCD4 were subjected to Western blotting using anti-FLAG and anti-His6 antibodies and to mass spectrometry (Structural Genomics Consortium, University of Oxford, UK) to confirm primary sequence fidelity.
Blue native PAGE (BN-PAGE)
Blue native PAGE was performed using 4–20% polyacrylamide gradient gels generated in-house. The buffering components of the gel included 50 mM Bis-tris and 500 mM aminocaproic acid, pH 7.0. Protein samples (8 μg) were incubated in the presence or absence of SDS (1%) for 10 min prior to loading. Samples and unstained protein standards (NativeMark; Life Technologies) were run in cold anode (50 mM Bis-tris pH 7.0) and deep blue cathode buffers (50 mM Tricine, 15 mM Bis-tris, 0.02% Coomassie G-250, pH 7.0) for 1 h at 100 V, whereupon the cathode buffer was replaced with slightly blue cathode buffer (50 mM Tricine, 15 mM Bis-tris, 0.002% Coomassie G-250, pH 7.0) and electrophoresis continued for 2–3 h at 180 V. Following electrophoresis, gels were stained with Coomassie brilliant blue R-250.
Chemical crosslinking
Prior to crosslinking, LMBD1 was diluted to 2.5 μM (0.15 mg/ml) and ABCD4 was diluted to 1.2 μM (0.08 mg/ml) in gel filtration buffer. For LMBD1 crosslinking, ultrapure EM grade glutaraldehyde (EM Sciences) was mixed with samples to a final concentration of 0.025% (w/w). Aliquots of LMBD1 were removed from the reaction mixture following incubation for 10, 30, and 60 min at 25 °C. To ensure specific crosslinking with glutaraldehyde, a control reaction of LMBD1 preincubated with 1% SDS followed by glutaraldehyde treatment (0.025%) for 1 h was performed concurrently. For ABCD4, formaldehyde (Pierce) was added to a final concentration of 0.075% (w/v) to initiate crosslinking; aliquots were removed following incubation for 30 min, 1 h, and 4 h at 25 °C. To terminate crosslinking for all reactions, aliquots were treated with SDS-PAGE sample buffer for 10 min prior to loading onto a 4–12% polyacrylamide gel prepared in-house. Following electrophoresis, crosslinked samples were visualized by silver staining.
Size exclusion chromatography – multi-angle light scattering (SEC-MALS)
For molecular mass determination of protein-detergent complexes (PDCs), size exclusion chromatography was coupled to multi-angle light scattering, refractive index, and UV detection. Both LMBD1 and ABCD4 were applied to a Superdex 200 10/300 GL column (GE Healthcare Life Sciences) connected to a Waters 2695 HPLC system at 20 °C at a flow rate of 0.25 ml/min using gel filtration buffer. Elution was monitored inline by absorption at 280 nm (Waters 2489 UV/Vis detector), by refractive index at 658.0 nm using an Optilab rEX detector (Wyatt Technology), and by multi-angle light scattering at 656.0 nm using the three detectors (49°, 90°, 131°) of the miniDAWN TREOS (Wyatt Technology). Data were analyzed using ASTRA 5.3.4.16 software (Wyatt Technology) using a standard template for determination of PDC molecular masses and the “protein conjugate” module for protein and detergent contributions to the PDC, according to Slotboom et al. (Citation2008). Rabbit muscle aldolase (GE Healthcare Life Sciences; 158 kDa) was used as internal standard for detector normalization. A (dn/dc)protein value of 0.192 ml/mg was used for LMBD1 and ABCD4 based on SEDFIT estimations (Schuck, Citation2000). The (dn/dc)detergent of DDM-CHS, 0.185 ml/mg, was calculated offline in batch mode using a standard curve of known detergent concentrations in gel filtration buffer. The extinction coefficient of LMBD1 at 280 nm (1.707 ml mg−1 cm−1) was calculated from primary sequence whereas the contribution of DDM-CHS at 280 nm was considered negligible (Slotboom et al., Citation2008).
Surface plasmon resonance (SPR)
Interactions between ABCD4 (310 kDa), LMBD1 (220 kDa), MMACHC (32 kDa), and/or MMADHC (32 kDa) were examined using label-free, real-time BIACORE 3000 instrumentation (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Experiments were performed on research-grade CM4 sensor chips at 25 °C using filtered (0.2 μm) and degassed HBS-ED running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.02% (w/v) DDM). Fatty acid free bovine serum albumin (BSA, 66 kDa) was from Sigma (#A8806), Pierce Gentle Elution (PGE) was from Thermo Scientific (#21027), and Anatrace detergents were from Affymetrix (DDM #D310 and Empigen #D350); maltose-binding protein (MBP, 42 kDa) was purified in-house and all other chemicals were reagent-grade quality.
Low- (∼100 RU), medium- (∼400 RU), and high-density (∼1000 RU) ABCD4-immobilized surfaces were prepared using the Biacore Amine Coupling kit (10 μg/ml ABCD4 in 10 mM sodium acetate pH 5.0 containing 0.02% (w/v) DDM); corresponding reference surfaces were prepared in the absence of any ABCD4 protein. To examine binding affinity, LMBD1 (0–100 nM, 2-fold serial dilutions) was titrated to steady-state plateaus using low flow rates (10 μl/min × 10 min association + 20 min dissociation). To cross-validate our results in the reversed “ligand-analyte” orientation, amine-coupled LMBD1-immobilized surfaces were prepared and tested in the similar manner. MMACHC and MMADHC were also amine-coupled (200–700 RU each) in complementary experiments to test for their interaction with ABCD4 and LMBD1, as well as solution-phase MMACHC or MMADHC (positive controls). In all cases, sensor chip surfaces were regenerated between sample injections at 50 μl/min using two 30-sec pulses of solution I (PGE containing 0.05% (v/v) Empigen), solution II (1 M imidazole containing 0.05% (v/v) Empigen), solution III (PGE containing 33 mM H3PO4, 1.5 M NaCl, and 0.05% (v/v) Empigen), solution IV (50 mM NaOH containing 1 M NaCl and 0.05% (v/v) Empigen), and solution V (HBS-ED) followed by routine washing procedures (i.e. “extraclean” and “wash” options).
SPR data were doubled-referenced (Myszka, Citation1999) and are representative of triplicate injections acquired from at least three independent trials. For each replicate series, a buffer blank was injected first, the highest titrant concentration second, and serial dilutions followed (from the lowest to the highest concentration repeated); comparing responses between the two highest titrant injections verified consistent immobilized surface activity throughout each assay. Apparent equilibrium dissociation constants (KD) were determined by global fitting of the data (averaged responses at the end of each association phase [Req] plotted versus concentration) to a “steady-state affinity” model in the BIAevaluation software (v 4.1). In all cases, theoretical binding maxima were predicted using the following equation:
where Rmax is the maximal binding response (RU) at saturation; MA is the molecular mass (kDa) of the injected analyte; ML is the molecular mass (kDa) of the immobilized ligand; RL is the amount (RU) of immobilized ligand; and n is the predicted binding stoichiometry (e.g. 1:1).
Phage display
Phage display was performed for MMACHC as previously described (Plesa et al., Citation2011). Pair-wise alignments of affinity-selected peptides against MMACHC were performed against LMBD1 and ABCD4 using the REceptor LIgand Contacts (RELIC) server MATCH (Mandava et al., Citation2004) and the MatchScan program (Deme et al., Citation2012; Plesa et al., Citation2011).
Results
Purification of recombinant LMBD1 and ABCD4
Extensive construct and detergent screening for both LMBD1 and ABCD4 was performed in collaboration with the Integral Membrane Proteins (IMP) group of the Structural Genomics Consortium Oxford. Briefly, N- and C-terminal truncation constructs of LMBD1 and ABCD4 (20 each) were cloned into expression vectors with either a His6-tag at the N-terminus or a His10/FLAG-tag at the C-terminus. Selection of the deletion constructs was based on secondary structure, disorder, sequence alignments, and topology predictions. Baculovirus for all LMBD1 and ABCD4 constructs were generated and used to infect small-scale Sf9 cultures for purification screening. This involved extraction of all LMBD1 and ABCD4 constructs from infected Sf9 membranes in the standard detergent n-dodecyl-β-D-maltoside (DDM) followed by immobilized metal affinity chromatography and SDS-PAGE. While several constructs of LMBD1 and ABCD4 were identified as soluble using this technique, we selected the more physiologically-relevant full-length (not truncated) C-terminal His10/FLAG-tag constructs of LMBD1 and ABCD4 for detergent screening assays. These constructs were purified in 12 detergent conditions and tested for monodispersity using analytical size exclusion chromatography (aSEC). LMBD1 was monodisperse by aSEC in DDM and mixed DDM-cholesteryl hemisuccinate (CHS) micelles but decylmaltoside (DM), undecylmaltoside (UDM), and decyl maltose neopentyl glycol (DMNG) necessitated supplementation with cholesteryl hemisuccinate for monodispersity (data not shown). For ABCD4, solubilization in mixed DDM-cholesteryl hemisuccinate (CHS) micelles yielded non-aggregated protein by aSEC but was polydisperse (described below).
To obtain sufficient material for biophysical studies, full-length constructs of LMBD1 and ABCD4 (incorporating tandem C-terminal affinity tags, His10-FLAG) were expressed and purified from large-scale Sf9 cultures (4 l each) using baculovirus generated from small-scale screening. LMBD1 and ABCD4 were extracted from Sf9 membranes by solubilization in DDM plus CHS (DDM-CHS) and subjected to Ni-NTA affinity chromatography (). Affinity-purified LMBD1 (>95% pure) exhibited smearing by SDS-PAGE, indicative of the six N-glycosylation sites that are predicted (Rutsch et al., Citation2009). Smearing was eliminated by adding Peptide-N-Glycosidase F (PNGase F) to samples prior to electrophoresis. Following affinity tag removal and extensive deglycosylation, LMBD1 migrated as two distinct species (55 kDa and 57 kDa). Continued SDS-PAGE heterogeneity was attributed to the incomplete deglycosylation of LMBD1 by PNGase F; only the 55 kDa band was visualized following exogenous PNGase F addition. These results indicated that PNGase F activity in SDS is maintained and yielded complete deglycosylation only when LMBD1 was denatured, i.e. deglycosylated LMBD1 migrated to an apparent molecular weight of 55 kDa, whereas partially deglycosylated LMBD1 migrated to 57 kDa. Based on the marginal difference in mass, we propose that partially deglycosylated LMBD1 is detected because of a single N-glycosylation site that is inaccessible to PNGase F under native conditions; resistance to PNGase F treatment is a common outcome for detergent-solubilized membrane proteins (Boulter & Wang, Citation2001). Extending PNGase F incubation times, screening other buffer conditions, and testing the activity of alternate deglycosylating enzymes (such as endoglycosidase H) did not alleviate this heterogeneity. Regardless of the observed glycosylation heterogeneity, LMBD1 was homogeneous and monodisperse based on aSEC ().
Figure 1. Purified, detergent-solubilized LMBD1 and ABCD4. (A) Left, SDS-PAGE of recombinant LMBD1; lane M, molecular mass standards; lane 1, affinity-purified LMBD1; lane 2, affinity-purified LMBD1 plus PNGase F; lane 3, LMBD1 following proteolysis by TEV protease and deglycosylation by PNGase F; lane 4, cleaved LMBD1 following reverse affinity purification; lane 5, cleaved LMBD1 following reverse affinity purification plus exogenous PNGase F. Right, SDS-PAGE of recombinant ABCD4; lane M, molecular mass standards; lane 1, affinity-purified ABCD4; lane 2, ABCD4 following TEV proteolysis; lane 3, ABCD4 following reverse affinity purification; lane 4, ABCD4 following reverse purification plus exogenous PNGase F. (B) aSEC chromatogram of LMBD1 (black), ABCD4 (grey), and molecular weight standards (dashed; scaled to LMBD1 axis); T, thyroglobulin (670 kDa); G, γ-globulin (158 kDa); O, ovalbumin (44 kDa).
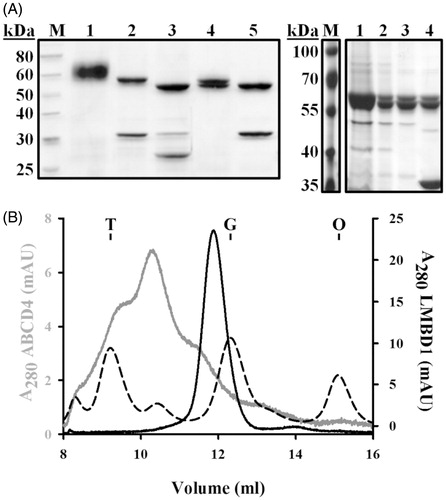
ABCD4 was purified using a similarly optimized workflow. Following preliminary Ni-NTA purification, ABCD4 migrated at a molecular mass of 62 kDa and was >95% pure by SDS-PAGE (). Following removal of the affinity tag, subsequent purification yielded two bands (62 kDa and 60 kDa). ABCD4 is not predicted to be glycosylated and, accordingly, we demonstrate that treatment with PNGase F does not shift its molecular mass by SDS-PAGE (). Alternatively, immunoblotting (anti-His10 or anti-FLAG) revealed that the 62 kDa band retained its affinity tag, whereas the 60 kDa did not, indicative of incomplete TEV proteolysis (data not shown). Extending TEV protease incubation times or altering the TEV:ABCD4 ratio did not reduce the presence of the 62 kDa band. The apparent molecular masses derived by SDS-PAGE for both LMBD1 and ABCD4 deviated from the theoretical primary sequence calculations, consistent with membrane proteins’ binding SDS to a greater degree than soluble proteins (Rath et al., Citation2009).
Purified LMBD1 and ABCD4 in DDM-CHS were subjected to aSEC to assess hydrodynamic properties of protein-detergent complexes (PDCs) (). LMBD1 was monodisperse in solution, yielding a peak corresponding to an apparent PDC molecular mass of 230 kDa based on molecular mass standards. ABCD4 yielded a polydisperse profile with three apparent peaks: ∼600 kDa minor leading peak, ∼350 kDa major peak, and ∼200–250 kDa minor trailing peak. Purification of an ABCD4 construct with an N-terminal affinity tag demonstrated increased susceptibility to TEV protease but yielded a similar polydisperse aSEC profile (data not shown), indicating that the heterogeneity of recombinant ABCD4 is not influenced by the location of the affinity tag but may be an intrinsic property of the membrane protein itself.
Oligomerization of LMBD1 and ABCD4
Due to inherent difficulties in elucidating membrane protein oligomerization by aSEC, alternative techniques were adopted. Blue native PAGE (BN-PAGE) assesses membrane protein complexes by native electrophoresis such that negatively charged Coomassie G-250 exchanges with the protein-bound detergent; the membrane protein thus migrates through the electric field. This technique has widely been utilized in combination with SDS-mediated complex dissociation to analyze purified components of bacterial secretion systems (Duong, Citation2003; Maillard et al., Citation2007; van der Does et al., Citation2003). LMBD1 subjected to BN-PAGE yielded a homogeneous band with apparent molecular mass of 180–200 kDa when compared to a molecular weight ladder derived from soluble proteins (). Treatment of LMBD1 with SDS prior to electrophoresis resulted in a shift to an apparent molecular mass of 90–100 kDa. While elucidation of the molecular masses of membrane protein complexes using soluble standards is inherently difficult due to the differential Coomassie-binding properties of soluble and membrane proteins, our results suggest that subunit dissociation through SDS treatment yields monomeric LMBD1 and that native LMBD1 is dimeric. Similar to its aSEC profiles, BN-PAGE profiles of ABCD4 were heterogeneous, with visible bands corresponding to molecular masses of ∼300 and ∼600 kDa (). SDS-treated ABCD4 migrated to a single species with apparent molecular mass of 90 kDa, again suggesting that ABCD4 exists as a higher-order oligomeric complex.
Figure 2. Self-association of LMBD1 and ABCD4. (A) BN-PAGE of purified LMBD1 (left) and ABCD4 (right). Lanes 1, DDMCHS-solubilized; lanes 2, SDS-solubilized (1%); lanes M, native molecular mass markers. (B) Chemical crosslinking of LMBD1 with glutaraldehyde (0.025%) at 25 °C for 10 min (lane 3), 30 min (lane 4), and 1 h (lane 5). Untreated LMBD1 (lane 1) or a 1 h glutaraldehyde-treated LMBD1 sample containing 1% SDS (lane 2) were used as controls. (C) Chemical crosslinking of ABCD4 with formaldehyde (0.1%) at 25 °C for 30 min (lane 2), 1 h (lane 3) and 4 h (lane 4). Untreated ABCD4 (lane 1) used as control. (D) SEC-MALS analysis of LMBD1 (solid) and ABCD4 (dashed), with absorbance (280 nm) profiles displayed and plotted against molar masses. The three major peaks of ABCD4, corresponding to PDCs, are indicated as red dots. For LMBD1, molecular mass contributions of protein (blue) and detergent (yellow) were deduced from the molecular mass of the PDC (green) using the ASTRA method (Slotboom et al., Citation2008).
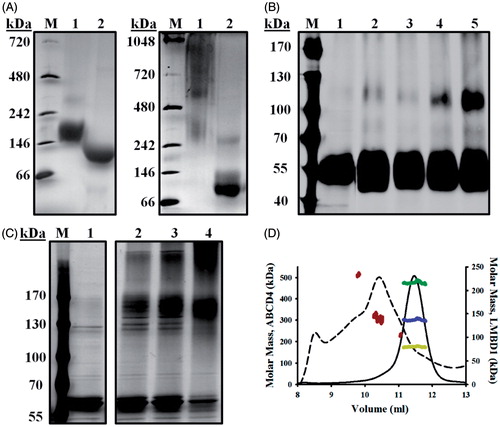
To further assess oligomerization, chemical crosslinking was performed on purified LMBD1 or ABCD4 followed by SDS-PAGE and visualization with silver staining. Untreated LMBD1 migrated to a molecular mass of 55 kDa, whereas 1 h treatment of LMBD1 with glutaraldehyde (0.025%) resulted in the formation of a protein adduct with molecular mass of 110 kDa, consistent with dimeric LMBD1 (). To ascertain that this adduct did not arise from non-specific crosslinking, LMBD1 was treated with both SDS and glutaraldehyde, effectively preventing dimer formation. For ABCD4, treatment with formaldehyde (0.1%) over 4 h resulted in the formation of complexes consistent in mass with dimeric ABCD4 (120–150 kDa) and a higher molecular weight oligomer, compared to monomeric molecular masses of 60 and 62 kDa in untreated samples (). The dimeric form predominated at 30 min and 1 h, with the higher molecular weight oligomer occurring only after extended incubation (4 h).
Building upon our BN-PAGE and crosslinking analyses, we then employed size exclusion chromatography with multi-angle light scattering (SEC-MALS). SEC-MALS allows for direct molecular mass determination of PDCs (including contributions arising from the individual protein or detergent components) and does not require a calibration curve composed of molecular mass standards (Slotboom et al., Citation2008). Analysis of LMBD1 revealed a molecular mass of 220 kDa for the PDC (), consistent with the 230 kDa estimation from our aSEC analyses (); the LMBD1 protein component contributed 135 kDa, whereas the protein-bound detergent (DDM-CHS) contributed 85 kDa. Given the theoretical molecular mass of deglycosylated LMBD1 is 62.2 kDa, our SEC-MALS analyses indicated that LMBD1 was dimeric. While the heterogeneity of ABCD4 precluded estimates of individual protein/detergent contributions to the PDC, we confirmed molecular masses of the three prominent PDCs observed in our aSEC profiles: a minor leading peak with molecular mass of 510 kDa, a minor trailing peak of 240 kDa and the prominent peak corresponding to 310 kDa (). Based on the theoretical molecular mass of ABCD4 (69.5 kDa) and our chemical crosslinking results, we propose that this major 310 kDa peak represents homodimeric ABCD4. Our findings are consistent with literature reports indicating that ABC half transporters dimerize to reconstitute functional transporters (Hillebrand et al., Citation2007; Liu et al., Citation1999). While ABC half transporters have also been shown to tetramerize (Xu et al., Citation2004), we contend that the protein contribution of tetrameric ABCD4 (280 kDa) to the 310 kDa PDC would be too substantial to permit ABCD4 solubilization based upon the minor amount of bound detergent (30 kDa). Overall, our results suggest that the leading shoulder (500–600 kDa) is tetrameric ABCD4, and the trailing shoulder (220–240 kDa) is monomeric ABCD4.
LMBD1 and ABCD4 interact by surface plasmon resonance (SPR)
Surface plasmon resonance was employed to test for the interaction between both membrane proteins. Initially, single-cycle titrations detected specific, dose-dependent binding between LMBD1 (0–100 nM) and amine-coupled ABCD4 (Supplementary Figure 1A, available online); in comparison, little or no signal changes were observed with significantly higher concentrations of MBP (0–400 nM) or BSA (0–600 nM) titrated under similar conditions. While saturable LMBD1-ABCD4 binding was also detectable using multi-cycle titrations in the reversed “ligand-analyte” orientation (Supplementary Figure 1B–C), it was easier to regenerate sensor chip surfaces when LMBD1 was flowed over immobilized ABCD4. Hence, this orientation was adopted for subsequent experiments.
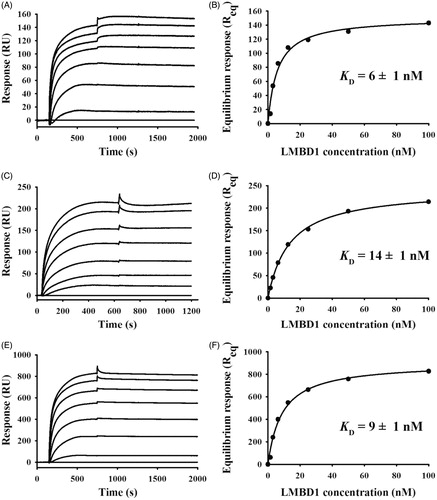
To further analyze the kinetics of the LMBD1-ABCD4 interaction, LMBD1 was titrated over low-, medium-, and high-density amine-coupled ABCD4 surfaces (, respectively). Immobilized ABCD4 was functional in all cases (i.e. surface activities >80%) and the saturable, dose-dependent LMBD1 binding responses correlated well with a 1:1 PDC:PDC stoichiometry (based upon the PDC molecular masses calculated by SEC-MALS, i.e. 220 kDa for LMBD1 and 310 kDa for ABCD4). Slow dissociation rates (<10%) did not permit fitting of the data using the simple 1:1 kinetics model. Alternatively, the multi-density titrations were analyzed according to the steady-state amounts of LMBD1 bound at the end of each association phase (, respectively); importantly, the acquired binding isotherms and low nanomolar KD values were similar in either “analyte-ligand” orientation (see Supplementary Figure 1C for comparison). Independent of mass transport effects, steady-state analyses for LMBD1 over all ABCD4 surfaces were unequivocal in terms of: (i) specific, dose-dependent binding characterized by slow dissociation kinetics; and (ii) low nanomolar binding affinities. Taken together, our SPR data indicate that the specific, dose-dependent interaction between LMBD1 and ABCD4 is stable (i.e. slow dissociation rate kinetics) and high-affinity (i.e. apparent equilibrium dissociation constant (KD) of ∼10 nM; n = 7).
Figure 3. Representative SPR kinetics of LMBD1 binding to amine-coupled ABCD4 at variable surface densities. Kinetics of LMBD1 (0–100 nM, 2-fold serial) binding to low (A, 125 RU), medium (C, 350 RU), and high (E, 1150 RU) density ABCD4 surfaces in multi-cycle mode (25 µl/min × 10 min association + 20 min dissociation). Corresponding isotherms (B, D, F) in which steady-state binding responses were plotted versus analyte concentrations (circles); fits to the “steady-state” affinity model in BIAevaluation (lines) predict low nanomolar KD for LMBD1-ABCD4 interaction with 1:1 stoichiometry overall.
Identification of potential MMACHC-binding sites on LMBD1 and on ABCD4 by phage display
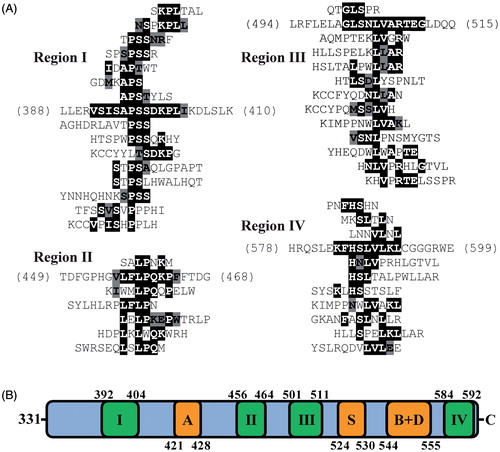
Phage display was used to predict regions on LMBD1 that may interact with MMACHC. Ensembles of 129 Ph.D.-C7C peptides and 149 Ph.D.-12 peptides previously isolated from MMACHC-binding phages (Plesa et al., Citation2011) were aligned against the primary sequence of LMBD1, using RELIC/MATCH and MatchScan. Thirty-four MMACHC-affinity selected peptides aligned to cytoplasmic loops (Supplementary Table 1, available online) based on the proposed topology of LMBD1 (UniProt: Q9NUN5), producing three distinct clusters (). Eight peptides aligned to residues 135–145 (region I), the loop between TM helices 3 and 4. The largest cluster consisted of 20 aligned peptides (region II), aligning to residues 215–225 within the largest cytoplasmic segment of LMBD1 between TM5 and TM6. The remaining six peptides mapped within this same loop but to residues 260–267 (region III).
Figure 4. Phage display identifies putative MMACHC binding sites on LMBD1. (A) MMACHC affinity-selected peptides aligned to three regions within cytoplasmic loops of LMBD1 (I–III). Exact sequence matches are highlighted in black; conservative matches are highlighted in grey. (B) Topology diagram of TM helices 3–6 of LMBD1 using boundaries from UniProt (Q9NUN5) and visualization with TOPO2. MMACHC-binding regions displayed in blue and denoted by roman numerals.
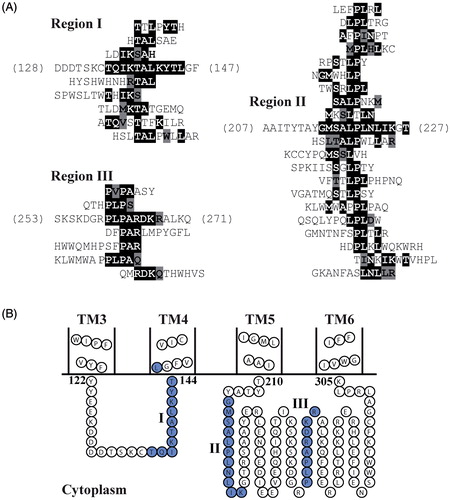
The same procedure was used to predict regions on ABCD4 that may interact with MMACHC. Affinity-selected peptides against MMACHC (Plesa et al., Citation2011) were mapped to the primary sequence of ABCD4. Forty-three affinity-selected peptides aligned to ABCD4 and formed distinct clusters (; Supplementary Table 2). All four predicted MMACHC-binding regions (I–IV) localized to the proposed cytoplasmic C-terminal NBD of ABCD4 (UniProt: O14678). Region I (residues 392–404) was N-terminal to the proposed Walker A motif (residues 421–428). Regions II (residues 456–464) and III (residues 501–511) mapped between the Walker A motif and proposed ABC signature (residues 524–530), Walker B motifs (544–549), and D-loop (548–555). The final region (IV; residues 584–592) mapped to the C-terminus of the NBD.
Figure 5. Phage display identifies putative MMACHC binding sites on ABCD4. (A) MMACHC affinity-selected peptides aligned to four regions within the C-terminal NBD of ABCD4 (I–IV). Exact sequence matches are highlighted in black; conservative matches are highlighted in grey. (B) Linear representation of the NBD of ABCD4 with proposed catalytic motifs shown in orange: A, Walker A; S, ABC signature; B + D, Walker B and D-loop. MMACHC-binding regions are annotated as roman numerals and displayed in green.
Binary interactions of LMBD1 or ABCD4 with MMACHC or MMADHC by SPR
To corroborate our phage display predictions, additional SPR analyses were performed in which MMACHC and MMADHC (32 kDa each) were immobilized, thereby maximizing the signal range with the significantly larger LMBD1 (220 kDa) and ABCD4 (310 kDa) membrane proteins. Fixed concentration injections of LMBD1 and ABCD4 (100 nM each) yielded specific binding responses (1500–2000 RU) against immobilized MMACHC as compared to BSA (50 RU; ); even with normalization for the 3- to 5-fold difference in molecular mass between the PDCs and BSA, the binding responses with LMBD1 and ABCD4 were still significantly larger compared to BSA (e.g. 50 RU observed, as compared to 4-fold scale up = ∼200 RU). In contrast, LMBD1 and ABCD4 elicited only minor binding responses (20–30 RU) against immobilized MMADHC as compared to BSA (5 RU; 4-fold scale up = ∼20 RU; ). Even with normalization for the 3.5-fold difference in the amine-coupled densities between MMACHC (700 RU) and MMADHC (200 RU), the LMBD1 and ABCD4 binding responses were still extremely low with MMADHC when compared to the theoretical Rmax values (signal range of 1300–1900 RU anticipated, as readily achieved with MMACHC). While these results are not suggestive of a role for MMADHC interacting with LMBD1 and ABCD4, we cross-validated the functional integrity of our cytoplasmic protein preparations by titrating MMACHC with MMADHC (Supplementary Figure 2A); the acquired binding profiles were consistent with our previous publications (thiol-coupled MMACHC, Plesa et al., Citation2011; amine-coupled MMADHC, Deme et al., Citation2012).
Figure 6. Representative SPR for LMBD1 and ABCD4 binding to amine-coupled MMACHC and MMADHC. Kinetics of buffer (black), BSA (red), ABCD4 (green), or LMBD1 (blue), each at 100 nM, binding to amine-coupled MMACHC (A, 700 RU) or MMADHC (B, 200 RU) at 25 μl/min (1 min association + 5 min dissociation). Multi-cycle titrations of ABCD4 (C) and LMBD1 (E) binding (0–100 nM, all 2-fold serial) to immobilized MMACHC (700 RU) at 25 μl/min (10 min association + 20 min dissociation). Corresponding binding isotherms in which steady-state binding responses were plotted versus analyte concentrations (circles; D, ABCD4; F, LMBD1) and fit to the “steady-state affinity” model in BIAevaluation (lines).
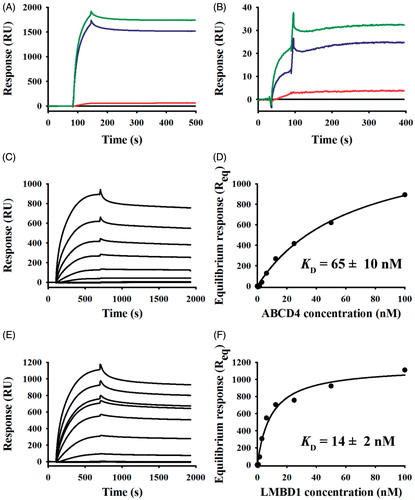
To examine their binding specificity with MMACHC in greater detail, detergent-solubilized ABCD4 () and LMBD1 () were titrated over amine-coupled MMACHC surfaces. ABCD4 and LMBD1 exhibited saturable, dose-dependent binding to MMACHC and similar affinities were detected (KD of 65 ± 10 nM versus 14 ± 2 nM, respectively; n = 3 for each). As a positive control, we also titrated MMACHC over the same immobilized MMACHC surfaces (Supplementary Figure 2B); similar to our previous publication (Deme et al., Citation2012), the association phase deviated from simple 1:1 kinetics (i.e. multi-phasic MMACHC-MMACHC interaction).
Discussion
The molecular bases of inborn cblF and cblJ defects in vitamin B12 metabolism were established when LMBRD1 and ABCD4 were identified to encode putative lysosomal exporters of Cbl (Coelho et al., Citation2012; Rutsch et al., Citation2009). Mutations in their encoded gene products, LMBD1 and ABCD4 respectively, prevent the lysosomal release of Cbl to the cytoplasm. Our current studies extend these findings by reporting the recombinant production of LMBD1 and ABCD4; even with our optimized protocol, the overall yields of these membrane proteins were limited (∼100 μg/l of culture) but both were highly pure (>95%) and stable in solution – essential criteria for structural and biophysical analyses.
Because of limited structure-function studies that have been published to date, our understanding of LMBD1 and ABCD4 has been primarily derived from homologous proteins. LMBD1 shares homology with the lipocalin-1 interacting membrane receptor (LIMR). LIMR is widely expressed across tissues and localizes to the plasma membrane (Wojnar et al., Citation2001), in contrast to LMBD1 which demonstrates only minor localization to the plasma membrane (Tseng et al., Citation2013). Similar to LMBD1, LIMR contains nine putative TM helices. LIMR binds and internalizes lipocalins via receptor-mediated endocytosis. Recombinant production of human LIMR in a Drosophila expression system was recently demonstrated by Hesselink and colleagues (Hesselink & Findlay, Citation2013). Congruent with our LMBD1 data, the authors demonstrated that DDM-solubilized LIMR forms dimers in solution. Our BN-PAGE, crosslinking, and SEC-MALS results for LMBD1 are consistent with those for LIMR, in that LMBD1 predominantly forms dimers in solution.
Recombinant ABCD4 demonstrates heterogeneity in its oligomeric state, but our results indicate that dimeric ABCD4 is the predominant form when solubilized in mixed DDM and CHS micelles. ABCD4 belongs to the D subfamily of peroxisomal ABC half transporters and shares homology with the other members: ABCD1, ABCD2, and ABCD3 (Wanders et al., Citation2007). Interaction analyses performed on other members of this subfamily support our conclusions that ABCD4 forms homodimers. Using yeast two-hybrid approaches, Liu and co-workers (Liu et al., Citation1999) showed that the cytoplasmic C-terminal NBD of ABCD1, ABCD2, and ABCD3 mediates homo- and heterodimerization in all possible combinations. In another study, ABCD1 and ABCD3, isolated from mouse liver, formed homodimers (Guimaraes et al., Citation2004). Live cell FRET microscopy identified homodimerization of ABCD1 and ABCD3 in addition to the formation of an ABCD1-ABCD3 heterodimeric complex (Hillebrand et al., Citation2007).
Our SPR analyses provided direct evidence for binding between LMBD1 and ABCD4. Given that previous SPR literature indicated similar outcomes using direct or indirect immobilization of membrane proteins (Vigonsky et al., Citation2013), we chose direct amine-coupling (over indirect Ni-NTA capture) to minimize sample consumption. The high-affinity interaction that we detected between ABCD4 and LMBD1 (i.e. KD ∼10 nM by SPR) was characterized by a slow dissociation rate. Accordingly, stable complex formation between ABCD4 and LMBD1 may be an important step for mediating intracellular vitamin B12 transport. In the context of Cbl transport, the Escherichia coli ABC transporter BtuC2D2 mediates Cbl import into the cytoplasm of E. coli by forming a complex with the periplasmic Cbl-binding protein BtuF. On its own, BtuC2D2 has poor affinity for Cbl which necessitates the delivery of Cbl by BtuF. Similar to our ABCD4-LMBD1 outcomes, BtuF binds BtuC2D2 with high affinity and their interaction is characterized by a slow dissociation rate constant using SPR (Lewinson et al., Citation2010). Slow dissociation may therefore be an inherent property of interactions involved in the early transport of Cbl, given that we also observed slow dissociation when characterizing the MMACHC-MMADHC interaction (Deme et al., Citation2012; Plesa et al., Citation2011). To date, a BtuF-like lysosomal Cbl binding protein has not yet been identified in mammals; significantly, cultured cblF or cblJ fibroblasts accumulate non-protein bound Cbl, which argues against the presence of a lysosomal Cbl-binding protein. Instead, we propose that Cbl transport across the lysosome is facilitated by a high affinity complex formed between LMBD1 and ABCD4.
Our phage display predictions indicate that cytoplasmic regions of LMBD1 and ABCD4 can recruit MMACHC. We identified three cytoplasmic loop regions between TM helices 3–4 and 5–6 on LMBD1. There are no known point mutations within these regions in patients with the cblF defect. Rather, all known mutations in LMBRD1 result in protein truncation (Armour et al., Citation2013; Miousse et al., Citation2011; Rutsch et al., Citation2009). For ABCD4, we identified four regions within the cytoplasmic C-terminal NBD. These regions did not overlap with the putative motifs required for ATP hydrolysis, but two overlapped with known mutations in ABCD4 (p.G443_S485del and p.E583LfsX9) that would effectively abrogate the proposed binding residues. The predictive power of phage display is illustrated in our previous work where we proposed binding interfaces between MMACHC and MMADHC and then performed SPR (in vitro) and bacterial two hybrid (in vivo) analyses to provide direct evidence for their interaction (Deme et al., Citation2012; Plesa et al., Citation2011). Five proposed MMACHC-binding regions were mapped to residues C-terminal to Glu142 on MMADHC (Plesa et al., Citation2011). An independent group (Gherasim et al., Citation2013a) recently corroborated our findings by demonstrating that MMACHC can bind to an MMADHC variant lacking 115 N-terminal residues.
We now communicate phage display results that predict MMACHC-binding sites on ABCD4 and LMBD1. Our current SPR analyses provide unequivocal, direct evidence that both of these membrane proteins can interact with MMACHC in a specific, dose-dependent manner. While parallel analyses do not support MMADHC as a partner protein for ABCD4 or LMBD1, we performed control titrations to cross-validate our previously published SPR data for MMACHC-MMACHC and MMACHC-MMADHC (Deme et al., Citation2012; Plesa et al., Citation2011). Given that the functional integrity of our MMACHC and MMADHC preparations was intact, it is interesting to note that the affinity of MMACHC for ABCD4 or LMBD1 (KD ∼65 nM and KD ∼14 nM, respectively) is stronger compared to MMACHC for MMADHC (KD ∼360 nM). Thus, the interaction of MMACHC with these lysosomal exporters would be favoured over cytoplasmic MMADHC should they be mutually exclusive.
Our evidence for the recruitment of MMACHC to the cytoplasmic side of lysosomal exporters agrees with recent proposals (Froese & Gravel, Citation2010; Rutsch et al., Citation2011). Such proposals argue that MMACHC recruitment allows the vectorial delivery of Cbl following transport across the lysosomal membrane, preventing dilution and exposure of the reactive cofactor to the cellular milieu. In the cytoplasm, MMACHC catalyzes the conversion of “base-on” Cbl to the “base-off” state; however, the reaction rate is slow (∼20 min) (Koutmos et al., Citation2011). It has been proposed that the acidic environment of the lysosomal lumen promotes the “base-off” state and that this Cbl conformation is maintained following lysosomal export by binding directly to MMACHC, effectively circumventing the slow MMACHC-catalyzed reaction (Banerjee, Citation2006; Gherasim et al., Citation2013a). While LMBD1 is speculated to form the transport channel for this process (Coelho et al., Citation2012), we cannot discount that MMACHC interacts with both putative exporters given the similar binding affinities detected. Cbl binding to MMACHC may trigger dissociation from LMBD1/ABCD4 and subsequent recruitment to MMADHC; the Cbl-dependence of the MMACHC-MMADHC interaction has recently been reported (Gherasim et al., Citation2013a).
Conclusions
Results from this study are consistent with the channeling and chaperoning of Cbl that is mediated through protein–protein interactions (Banerjee, Citation2006; Gherasim et al., Citation2013b). To date, research on LMBD1 and ABCD4 has been mostly limited to studies in cultured cells. For the first time, our study describes the recombinant production of LMBD1 and ABCD4 and protein complexes formed between these lysosomal membrane proteins and with MMACHC. Our results provide the framework for functional assays that will provide insight into the activity of LMBD1 and ABCD4, thus advancing our basic understanding of early intracellular vitamin B12 metabolism.
Acknowledgements
The authors wish to thank the following colleagues at the Structural Genomics Consortium, Oxford: Leela Shrestha for baculovirus construction and initial detergent screening of LMBD1, Rod Chalk and Georgina Berridge for mass spectrometry services, and Shubhashish Mukhopadhyay for cell culture expression and training.
Funding
Research was supported by operating grants to J.W.C. from the Natural Sciences and Engineering Research Council [NSERC; RGPIN 7289-06-07] and to D.S.R. from the Canadian Institutes of Health Research [CIHR; MOP-15078]. The McGill SPR Facility thanks the Canada Foundation for Innovation [CFI grant to Montreal Integrated Genomics Group for Research on Infectious Pathogens] and Canadian Institutes of Health Research [CIHR Research Resource Grant 200610PRG-165657-PRG-CFAA-11449] for infrastructure support. The Structural Genomics Consortium (SGC) is a registered charity [no. 1097737] that receives funds from AbbVie, Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Eli Lilly Canada, the Novartis Research Foundation, Pfizer, Takeda, Canada Foundation for Innovation, the Ontario Ministry of Economic Development and Innovation, and the Wellcome Trust [092809/Z/10/Z].
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Supplementary material available online
Supplementary Figures 1 and 2
Supplementary Tables 1 and 2.
Supplementary Material
Download PDF (148.9 KB)References
- Armour CM, Brebner A, Watkins D, Geraghty MT, Chan A, Rosenblatt DS. 2013. A patient with an inborn error of vitamin B12 metabolism (cblF) detected by newborn screening. Pediatrics 132:e257–e261
- Banerjee R. 2006. B12 trafficking in mammals: A for coenzyme escort service. ACS Chem Biol 1:149–159
- Boulter JM, Wang DN. 2001. Purification and characterization of human erythrocyte glucose transporter in decylmaltoside detergent solution. Protein Expr Purif 22:337–348
- Chapel A, Kieffer-Jaquinod S, Sagne C, Verdon Q, Ivaldi C, Mellal M, et al. 2013. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 12:1572–1588
- Coelho D, Kim JC, Miousse IR, Fung S, Du Moulin M, Buers I, et al. 2012. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet 44:1152–1155
- Deme JC, Miousse IR, Plesa M, Kim JC, Hancock MA, Mah W, et al. 2012. Structural features of recombinant MMADHC isoforms and their interactions with MMACHC, proteins of mammalian vitamin B12 metabolism. Mol Genet Metab 107:352–362
- Duong F. 2003. Binding, activation and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J 22:4375–4384
- Froese DS, Gravel RA. 2010. Genetic disorders of vitamin B12 metabolism: Eight complementation groups – eight genes. Expert Rev Mol Med 12:e37
- Gherasim C, Hannibal L, Rajagopalan D, Jacobsen DW, Banerjee R. 2013a. The C-terminal domain of CblD interacts with CblC and influences intracellular cobalamin partitioning. Biochimie 95:1023–1032
- Gherasim C, Lofgren M, Banerjee R. 2013b. Navigating the B12 road: Assimilation, delivery, and disorders of cobalamin. J Biol Chem 288:13186–13193
- Guimaraes CP, Domingues P, Aubourg P, Fouquet F, Pujol A, Jimenez-Sanchez G, et al. 2004. Mouse liver PMP70 and ALDP: Homomeric interactions prevail in vivo. Biochim Biophys Acta 1689:235–243
- Hesselink RW, Findlay JB. 2013. Expression, characterization and ligand specificity of lipocalin-1 interacting membrane receptor (LIMR). Mol Membr Biol 30:327–337
- Hillebrand M, Verrier SE, Ohlenbusch A, Schafer A, Soling HD, Wouters FS, Gartner J. 2007. Live cell FRET microscopy: Homo- and heterodimerization of two human peroxisomal ABC transporters, the adrenoleukodystrophy protein (ALDP, ABCD1) and PMP70 (ABCD3). J Biol Chem 282:26997–27005
- Hodgkin DC, Kamper J, Mackay M, Pickworth J, Trueblood KN, White JG. 1956. Structure of vitamin B12. Nature 178:64–66
- Holzinger A, Kammerer S, Roscher AA. 1997. Primary structure of human PMP69, a putative peroxisomal ABC-transporter. Biochem Biophys Res Commun 237:152–157
- Kashiwayama Y, Seki M, Yasui A, Murasaki Y, Morita M, Yamashita Y, et al. 2009. 70-kDa peroxisomal membrane protein related protein (P70R/ABCD4) localizes to endoplasmic reticulum not peroxisomes, and NH2-terminal hydrophobic property determines the subcellular localization of ABC subfamily D proteins. Exp Cell Res 315:190–205
- Kim JC, Lee NC, Hwu PW, Chien YH, Fahiminiya S, Majewski J, et al. 2012. Late onset of symptoms in an atypical patient with the cblJ inborn error of vitamin B12 metabolism: Diagnosis and novel mutation revealed by exome sequencing. Mol Genet Metab 107:664–668
- Koutmos M, Gherasim C, Smith JL, Banerjee R. 2011. Structural basis of multifunctionality in a vitamin B12-processing enzyme. J Biol Chem 286:29780–19787
- Lewinson O, Lee AT, Locher KP, Rees DC. 2010. A distinct mechanism for the ABC transporter BtuCD-BtuF revealed by the dynamics of complex formation. Nat Struct Mol Biol 17:332–338
- Liu LX, Janvier K, Berteaux-Lecellier V, Cartier N, Benarous R, Aubourg P. 1999. Homo- and heterodimerization of peroxisomal ATP-binding cassette half-transporters. J Biol Chem 274:32738–32743
- Maillard AP, Lalani S, Silva F, Belin D, Duong F. 2007. Deregulation of the SecYEG translocation channel upon removal of the plug domain. J Biol Chem 282:1281–1287
- Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. 2004. RELIC – a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics 4:1439–1460
- Miousse IR, Watkins D, Rosenblatt DS. 2011. Novel splice site mutations and a large deletion in three patients with the cblF inborn error of vitamin B12 metabolism. Mol Genet Metab 102:505–507
- Myszka DG. 1999. Improving biosensor analysis. J Mol Recognit 12:279–284
- Plesa M, Kim J, Paquette SG, Gagnon H, Ng-Thow-Hing C, Gibbs BF, et al. 2011. Interaction between MMACHC and MMADHC, two human proteins participating in intracellular vitamin B12 metabolism. Mol Genet Metab 102:139–148
- Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci USA 106:1760–1765
- Rosenblatt DS, Hosack A, Matiaszuk NV, Cooper BA, Laframboise R. 1985. Defect in vitamin B12 release from lysosomes: Newly described inborn error of vitamin B12 metabolism. Science 228:1319–1321
- Rutsch F, Gailus S, Miousse IR, Suormala T, Sagne C, Toliat MR, et al. 2009. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat Genet 41:234–239
- Rutsch F, Gailus S, Suormala T, Fowler B. 2011. LMBRD1: The gene for the cblF defect of vitamin B12 metabolism. J Inherit Metab Dis 34:121–126
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619
- Shani N, Jimenez-Sanchez G, Steel G, Dean M, Valle D. 1997. Identification of a fourth half ABC transporter in the human peroxisomal membrane. Hum Mol Genet 6:1925–1931
- Slotboom DJ, Duurkens RH, Olieman K, Erkens GB. 2008. Static light scattering to characterize membrane proteins in detergent solution. Methods 46:73–82
- Stubbe J. 1994. Binding site revealed of nature's most beautiful cofactor. Science 266:1663–1664
- Tseng LT, Lin CL, Tzen KY, Chang SC, Chang MF. 2013. LMBD1 serves as a specific adaptor for insulin receptor internalization. J Biol Chem 288:32424–32432
- van der Does C, De Keyzer J, Van Der Laan M, Driessen AJ. 2003. Reconstitution of purified bacterial preprotein translocase in liposomes. Methods Enzymol 372:86–98
- Vassiliadis A, Rosenblatt DS, Cooper BA, Bergeron JJ. 1991. Lysosomal cobalamin accumulation in fibroblasts from a patient with an inborn error of cobalamin metabolism (cblF complementation group): Visualization by electron microscope radioautography. Exp Cell Res 195:295–302
- Vigonsky E, Ovcharenko E, Lewinson O. 2013. Two molybdate/tungstate ABC transporters that interact very differently with their substrate binding proteins. Proc Natl Acad Sci USA 110:5440–5445
- Wanders RJ, Visser WF, Van Roermund CW, Kemp S, Waterham HR. 2007. The peroxisomal ABC transporter family. Pflugers Arch 453:719–734
- Watkins D, Rosenblatt DS. 1986. Failure of lysosomal release of vitamin B12: A new complementation group causing methylmalonic aciduria (cblF). Am J Hum Genet 39:404–408
- Wojnar P, Lechner M, Merschak P, Redl B. 2001. Molecular cloning of a novel lipocalin-1 interacting human cell membrane receptor using phage display. J Biol Chem 276:20206–20212
- Xu J, Liu Y, Yang Y, Bates S, Zhang JT. 2004. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem 279:19781–19789