
Abstract
The PapC usher, a β-barrel pore in the outer membrane of uropathogenic Escherichia coli, is used for assembly of the P pilus, a key virulence factor in bacterial colonization of human kidney cells. Each PapC protein is composed of a 24-stranded β-barrel channel, flanked by N- and C-terminal globular domains protruding into the periplasm, and occluded by a plug domain (PD). The PD is displaced from the channel towards the periplasm during pilus biogenesis, but the molecular mechanism for PD displacement remains unclear. Two structural features within the β-barrel, an α-helix and β5-6 hairpin loop, may play roles in controlling plug stabilization. Here we have tested clusters of residues at the interface of the plug, barrel, α-helix and hairpin, which participate in electrostatic networks. To assess the roles of these residues in plug stabilization, we used patch-clamp electrophysiology to compare the activity of wild-type and mutant PapC channels containing alanine substitutions at these sites. Mutations interrupting each of two salt bridge networks were relatively ineffective in disrupting plug stabilization. However, mutation of two pairs of arginines located at the inner and the outer surfaces of the PD resulted in an enhanced propensity for plug displacement. One arginine pair involved in a repulsive interaction between the linkers that tether the plug to the β-barrel was particularly sensitive to mutation. These results suggest that plug displacement, which is necessary for pilus assembly and translocation, may require a weakening of key electrostatic interactions between the plug linkers, and the plug and the α-helix.
Introduction
Pili (also known as fimbriae) are adhesive surface structures expressed by many different bacteria. Uropathogenic strains of Escherichia coli express pili to adhere to host uroepithelial cells at the onset of urinary tract infections, enabling both bladder and kidney infections. The adhesive property of pili is vital for establishing bacterial colonization, and animal studies have indicated that pili are essential to the disease process (Crepin et al., Citation2012). P pili, encoded by the pap system, are utilized by uropathogenic E. coli to colonize the kidney and cause acute pyelonephritis (Roberts et al., Citation1994). P pili are heteropolymeric fibers composed of six different subunits assembled in a precise stoichiometry and order (PapGDEFAH) (Fronzes et al., Citation2008; Thanassi et al., Citation2012). The distal subunit PapG functions as an adhesin and contains a lectin domain that mediates binding to Gal-α(1–4)-Gal-containing glycolipids on the host cell membrane (Lund et al., Citation1987). A chaperone-usher pathway is used for the assembly and secretion of these essential virulence factors at the cell surface (Busch & Waksman, Citation2012; Geibel & Waksman, Citation2014). The chaperone-usher pathway is a bi-component system consisting of a periplasmic chaperone and an outer membrane protein, the usher, which catalyses assembly of the pilus fiber and provides a conduit for export of the nascent pilus to the bacterial surface (Busch & Waksman, Citation2012). In the pap chaperone-usher system, the usher is PapC and the chaperone is PapD. After entry into the bacterial periplasm via the Sec general secretory pathway, individual pilus subunits associate with the PapD chaperone by a mechanism termed donor strand complementation (DSC), whereby the incomplete Ig-like fold of the pilus subunit is complemented by a β-strand provided by the chaperone (Sauer et al., Citation1999). Through DSC, the chaperone promotes the proper folding of each subunit into its correct configuration, and prevents premature subunit-subunit interactions from forming in the periplasm. Chaperone-subunit complexes are then targeted to the PapC usher in the outer membrane for pilus biogenesis and translocation. The usher catalyzes the exchange of chaperone-subunit for subunit-subunit interactions (Nishiyama et al., Citation2008; Sauer et al., Citation1999). This process, called donor strand exchange (DSE), occurs at the periplasmic side of the usher and effectively promotes the ordered growth of the pilus. The growing pilus fiber is secreted through the usher channel to reach the cell surface.
Structural studies of PapC, and its homolog FimD used in type 1 pilus biogenesis, have provided much insight into the mechanism of pilus assembly and translocation through the usher (Geibel et al., Citation2013; Phan et al., Citation2011; Remaut et al., Citation2008). The PapC and FimD ushers are large outer membrane proteins (∼90 kD) composed of five domains. The membrane-embedded translocation domain is flanked by large globular domains that face the periplasmic space: an N-terminal domain (NTD) and two C-terminal domains (CTD1 and CTD2) (Geibel & Waksman, Citation2014). The NTD and CTD have β-sandwich folds and are used as binding sites for the chaperone-subunit complexes at various stages during the active cycle of the usher (Eidam et al., Citation2008; Ford et al., Citation2010; Ng et al., Citation2004; Nishiyama et al., Citation2005). The translocation domain is a kidney-shape, 24 stranded β-barrel delineating a pore that is occluded by a plug domain (PD) (). The PD is a 76-residue segment between β-strands 6 and 7, that folds inside the barrel lumen as a 6-stranded β-sandwich and connects to the barrel via two linkers (P-linker 1 and P-linker 2) originating from the periplasmic face (Remaut et al., Citation2008). Also within the translocation domain are a hairpin loop between β-strands 5 and 6 (the β5–6 hairpin), which dips toward the barrel lumen, and the only α-helix in the translocation domain, which caps the β5–6 hairpin from the extracellular side. These two elements are believed to confer stability to the surrounding region, particularly to the plug domain, as their removal causes increased pore permeability (Volkan et al., Citation2013), channel opening (Mapingire et al., Citation2009) and decreased interactions between conserved residues (Farabella et al., Citation2014). Specific communities of amino acids in the α-helix and the β5–6 hairpin were proposed to function in the transmission of a putative allosteric signal that triggers plug displacement (Farabella et al., Citation2014).
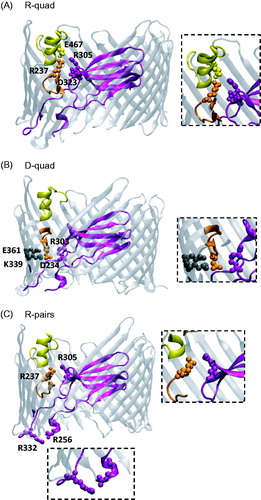
Figure 1. Cartoon-ribbon representations of PapC structure showing the locations of the mutated residues. (A) Salt-bridge network (R-quad) between residues R237, R305, D323 and E467, located at the interface between the PD (R305 and D323), hairpin (R237), and α-helix (E467). (B) Salt-bridge network (D-quad) between residues D234, R303, K339, and E361 located at the interface between the PD (R303), hairpin (D234), and barrel wall (K339 and E361). (C) Repulsive electrostatic interactions between the arginine pairs (R-pairs) R256-R332 on the two P-linkers, and R237-R305 between the PD and the hairpin loop. PapC structure from PDB 2VQI (Remaut et al., Citation2008) was rendered with the VMD program (Humphrey et al., Citation1996). Color code: Purple – PD; Orange – Hairpin; Yellow – α-helix; Gray – Barrel wall.
For assembly and translocation of the pilus fiber through the usher, the PD must be displaced. The crystal structure of a ternary complex of the FimD usher with the chaperone FimC delivering the first subunit FimH provides a snapshot of an early step in the translocation process (Phan et al., Citation2011). Comparison of this ternary complex with the structure of the inactive, plug-occluded usher shows that, upon usher activation, the PD swings towards the periplasmic side and the resulting open pore within the barrel becomes occupied by the lectin domain of FimH. The pilin domain of FimH remains associated with the chaperone, itself bound to the CTD of the usher. Biochemical and genetic studies indicate that chaperone-subunit complexes initially bind the usher NTD (Ng et al., Citation2004). Taken together, these data suggest a model of pilus assembly whereby the process starts with binding of the first chaperone-subunit complex to the usher NTD, followed by plug displacement and insertion of the first subunit in the vacant pore. Next, successive rounds of binding of incoming subunits to the NTD, handing over to the CTD, and linking to the last incorporated subunit through DSE extends the pilus fiber, concomitant with translocation of the fiber through the usher pore (Geibel & Waksman, Citation2014). While multiple lines of evidence indicate that the usher exists as dimeric or higher-order structures in the outer membrane, structural studies indicate that only a single pilus is assembled and translocated, with only one usher molecule providing the secretion pore (Li & Thanassi, Citation2009; Remaut et al., Citation2008). The usher oligomer may increase the efficiency of pilus assembly and provide an additional level of regulation (So & Thanassi, Citation2006; Werneburg et al., Citation2015).
Functional analyses are required to confirm structure-based hypotheses and obtain mechanistic details. Single-protein approaches can be particularly useful, and we initiated a functional study of PapC channel activity with the use of electrophysiology (Mapingire et al., Citation2009). While ion flux through the outer membrane is not the physiological function of PapC, the real-time measurement of such fluxes provides information on the dynamic states of the protein, and allows insights into the translocation process, as has been done for other bacterial pore-forming proteins (Delcour, Citation2013). In our previous study, electrical recordings were obtained from wildtype (WT) PapC and domain deletion mutants that had been purified and reconstituted into planar lipid bilayers. This domain analysis corroborated the structural evidence that the PD gates the channel shut, as the plugless mutant had a much more open phenotype than WT. Interestingly, the α-helix plays a role in keeping the PD lodged into the barrel lumen, as a PapC variant lacking this domain was prone to spontaneous large opening transitions of conductance similar to that of the plugless channel. Understanding the nature of the stabilization of the PD within the barrel lumen and the molecular mechanism for plug displacement is a crucial part of the overall comprehension of the usher molecular machine. Recent data point to the existence of an allosteric network of amino acids that may play a role in plug displacement (Farabella et al., Citation2014). In addition, several key residues that form several electrostatic networks that may serve to modulate the PD have been identified (Remaut et al., Citation2008). These networks are located at the interface of the PD with the barrel wall, α-helix, or β-hairpin loop. To assess the possible roles of these networks in plug stabilization, we conducted patch clamp experiments on PapC variants with mutations in residues participating in these electrostatic interactions. Our results suggest that repulsive interactions between the two linkers of the PD, and between the plug and the α-helix, play important roles in the modulation of usher gating.
Methods
Strains, media and chemicals
E. coli strain DH5α (Grant et al., Citation1990) was used for construction of PapC mutants, strain AAEC185 (Blomfield et al., Citation1991) was used for HA assays, and the multi-porin mutant BL21(DE3)Omp8 (Prilipov et al., Citation1998) was used for PapC purification. Bacteria were grown in LB medium containing 100 μg/ml ampicillin at 37 °C with aeration. For protein expression, PapC was induced at an OD600 of 0.6 for 2–2.5 h by addition of 0.1% L-arabinose. For protein purification, dodecyl-maltopyranoside (DDM) and lauryl(dimethyl)amine oxide (LDAO) were purchased from Anatrace. Thrombin was from Novagen. For electrophysiological experiments, the soybean phospholipids preparation ‘asolectin’ (L-α-Phosphatidylcholine Type II-S) was from Sigma, and the BioBeads used for reconstitution from Biorad. All other chemicals were from Sigma.
Construction and expression analysis of the PapC mutants
Plasmid pDG2, encoding WT PapC with a thrombin cleavage site and hexahistadine tag (His-tag) added to the C terminus, was previously described (Li et al., Citation2004). The PapC alanine substitution mutants were derived from pDG2 using QuikChange Site-Directed Mutagenesis (Stratagene) and the primers as listed in Supplementary Table 1 (available online). The PapC R237A (Farabella et al., Citation2014) and PapC R305A (Volkan et al., Citation2013) mutants were previously described. All PapC mutants were checked for proper construction by DNA sequencing. Each of the PapC mutants was compared with WT PapC for expression level in the outer membrane by inspection of Coomassie blue-stained SDS-PAGE gels and immunoblotting with anti-His-tag (Convance) antibody. Proper folding of the ushers in the outer membrane was checked by resistance to denaturation by SDS, as determined by heat-modifiable mobility on SDS-PAGE. These procedures were performed as described (Henderson et al., Citation2011).
Hemagglutination (HA) assay
The ability of each of the PapC mutants to complement a ΔpapC pap operon for assembly of P pili was determined by HA assay. Assays were performed by serial dilutions in microtiter plates as described (Henderson et al., Citation2011). Visual inspection was used to determine the HA titers as the highest-fold dilution of bacteria still able to agglutinate human red blood cells. Each mutant was analyzed in two independent assays, with each assay performed in triplicate, and the values were averaged.
Protein purification
The WT and mutant PapC proteins were purified from the bacterial outer membrane as previously described (Mapingire et al., Citation2009), except that the protein was not subjected to gel filtration chromatography. Instead, following removal of the His-tag by thrombin digestion, the protein was passed over a metal affinity column coupled to a benzamidine column, as described (Werneburg et al., Citation2015).
Patch-clamp electrophysiology
The pore-forming activity of reconstituted WT and mutant ushers were investigated with the patch-clamp technique, as described (Simonet et al., Citation2003). Pure proteins were reconstituted into asolectin at a protein:lipid ratio of 1:2000 to 1:3000 (w:w). Patch pipettes of ∼10 MΩ were filled with Buffer A (150 mM KCl, 10 μM CaCl2, 0.1 mM EDTA, 5 mM HEPES, pH 7.2). Patch clamp experiments were performed on unilamellar blisters emerging from liposomes when placed in Buffer B (Buffer A + 20 mM MgCl2). Pipettes were brought into contact with the blister membrane to generate a 1.0–10.0 GΩ seal. Patches were excised by brief air exposure, and the bath solution was subsequently exchanged for Buffer A, to obtain recordings in symmetric ionic conditions. Currents were recorded under voltage clamp, with an Axopatch-1D amplifier (Axon Instruments) and a CV-4 headstage. The currents were filtered at 1 KHz, digitized at 100 μs sampling intervals (ITC-18, Instrutech), and stored on a computer using the Acquire software (Bruxton). Control experiments performed on liposomes lacking reconstituted usher showed no channel activity. In less than 0.01% of the patches, nonspecific current deflections may be seen. Such patches are discarded.
Results
Electrostatic networks in the PapC usher and cellular phenotypes of the mutants
Examination of the PapC usher structure reveals the existence of groups of residues that are within distances supporting electrostatic interactions at the interface of the PD or its linkers and other structural elements in the usher, such as the β-barrel, the α-helix or the β5–6 hairpin (, ) (Remaut et al., Citation2008). Two of these groups include residues that appear linked by a network of salt bridges; one group involves R237, R305, D323 and E467, sustaining attractive interactions between residues of opposite charges located on the PD, the α-helix and the β5–6 hairpin ( and ); the other group contains D234, R303, K339 and E361, also sustaining attractive interactions between residues of opposite charges on the PD, the barrel wall and the β5–6 hairpin ( and ). Distances between interactive pairs of these networks are given in . A third group () is made of two pairs of same-charge residues (arginines) located at the periplasmic (R256-R332) and external (R237-R305) faces of the usher. Attractive interactions between the stacked parallel guanidinium groups of arginines have been reported when the guanidinium groups are in close proximity (<4 Å) (Vondrasek et al., Citation2009). In proteins with known crystal structures, there is ∼10% occurrence of guanidinium groups located within 6 Å of each other, and 40% of these are parallel (Lee et al., Citation2013). Here, the distances between the Cζ atoms of the guanidinium groups and the angle between the guanidinium groups are 6.9 Å and 50.1° for R256-R332, and 4.3 Å and 74.3° for R237-R305 ( lists the shortest distance between the N atoms of the guanidinium). Therefore, attractive forces between these arginine pairs are unlikely, and we postulate that the arginine pairs are involved in repulsive interactions. We hypothesized that these sets of electrostatic interactions might play a role in either stabilizing the PD within the barrel or aiding in PD expulsion to open the channel, and set out to test this hypothesis by constructing quadruple alanine mutants in each group. We will use the following nomenclature in the rest of the manuscript when referring to these mutants: R-quad mutant (R237A - R305A - D323A - E467A), D-quad mutant (D234A - R303A - K339A - E361A), and R-pairs mutant (R237A - R256A - R305A - R332A).
Table 1. Locations and distances between the charged functional groups of the residues in the three electrostatic networks investigated.
All of the PapC mutants expressed similarly to WT PapC. The PapC usher exhibits a characteristic heat modifiability in SDS-PAGE (Nakamura & Mizushima, Citation1976; Ng et al., Citation2004), in which the usher β-barrel domain remains folded in the absence of heating and migrates with faster mobility when compared to fully denatured (boiled) protein. Therefore, we used this shift in mobility as evidence of folding, and observed that none of the mutations affected the ability of the usher to form a stable β-barrel in the outer membrane (). A hemagglutination (HA) assay was used to assess the ability of each PapC mutant to assemble functional P pili on the bacterial surface. The HA titer for the D-quad mutant was the same as for WT PapC, whereas the R-quad mutant was deficient for pilus assembly (). The R-pairs mutant, however, was completely defective pilus assembly, with an HA titer of 0. Given the deficiencies of some of these mutants, we set out to analyze their functional properties at the molecular level by using single-channel electrophysiology.
Table 2. Folding, pilus expression, and average displacement voltage in WT and mutant PapC ushers.
Plug displacement in WT PapC
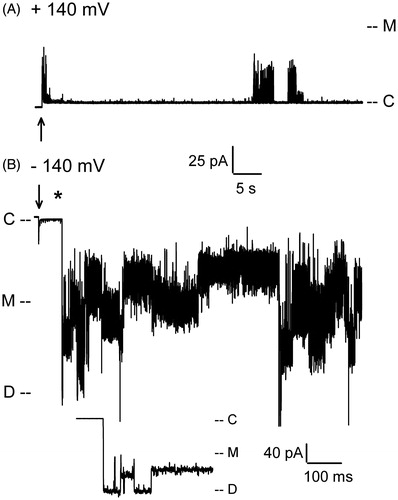
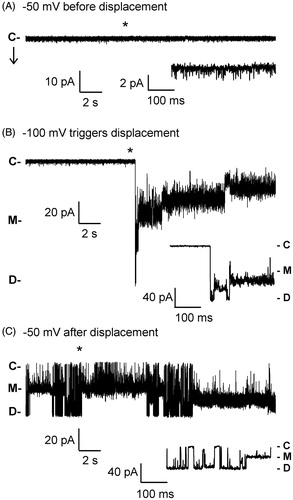
Our previous electrophysiological analyses of the WT PapC usher in planar lipid bilayer indicated that the channel reconstitutes as a dimer in the liposome membrane, and exists in a mostly closed conformation at relatively low membrane potentials (<100 mV) (Mapingire et al., Citation2009). However, the channel behaved as a dynamic entity, with frequent opening transitions of varying conductance. Many of these openings had a conductance lower than the monomeric conductance obtained from a plugless variant of the usher. These openings were interpreted as originating from small water-filled conduits at the barrel-plug interface that allow for leakage of ions and could be occasionally interrupted via occlusion by the NTD or CTD, or by movement of the plug or the barrel itself (Mapingire et al., Citation2009). Occasionally, large openings with monomeric conductance could be observed, likely representing the spontaneous displacement of the plug (Farabella et al., Citation2014; Mapingire et al., Citation2009); the frequency of these transitions increased with membrane potential (Farabella et al., Citation2014), but extremely high membrane potentials were never tested in these studies. We observed the same kinetics as in the previous studies using patch-clamp electrophysiology. At low membrane potentials of both polarities (<120–140 mV), the WT channel displays frequent transient openings of various sizes, ranging from ∼50 pS to ∼600 pS (150 mM KCl), a maximum value similar to the monomeric conductance of a plugless mutant in these conditions. A similar behavior is also seen at high positive membrane potentials (). However, high negative voltages (> ∼−140 mV) can trigger extremely large currents that can last for hundreds of seconds (). Initially, the observed conductance typically reaches a level of approximately ∼1.2 nS, corresponding to the dimeric conductance. This behavior is interpreted as the voltage-driven simultaneous displacements of both plugs from a dimeric usher; i.e., a plug displacement event. Interestingly, this plug displacement is never seen at positive voltages (), indicating not only the polarity dependence of the effect but also the reproducible unidirectional reconstitution of the usher.
Figure 2. Patch-clamp recordings of WT PapC activity at high membrane potentials. (A) At positive voltages, the channel shows a mixture of spontaneous transitions of small and large conductance. (B) The application of high negative voltage (−140 mV) triggers the simultaneous displacement of the plugs from both pores of the usher dimer. The asterisk (*) denotes the region that is expanded below the trace for better viewing of the short-lived transition to the dimeric current level. ‘C’ indicates the current level in the closed state of the usher, ‘M’ the monomeric level of conductance, and ‘D’ the dimeric level of conductance. The arrow marks the time at which the voltage was applied.
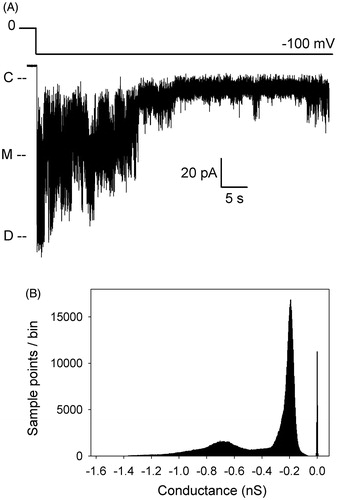
After displacement, the current quickly relaxes to lower values, albeit with a large amount of dynamic behavior indicative of a profound conformational flexibility of the usher after plug release. Amplitude histograms obtained from 10-sec recordings encompassing a plug displacement event and subsequent relaxation show that the usher samples a range of kinetic states with frequencies and dwell times that vary between experiments (Supplementary Figure 1, available online). Conductance levels are consistent across experiments, but the relative occupancies of these states, as reported by the height of the histogram peaks, are different from one displacement event to the next. A straightforward explanation for the current relaxation would be that it is due to plug re-entry into the barrel lumen. However, a similar behavior is found in a PapC mutant lacking the PD. As shown in , as soon as a relatively high voltage is applied to a patch containing a plugless channel, the trace jumps to a value corresponding to the dimeric open current, as expected for a channel lacking the plug. However, within a few hundreds of milliseconds, the trace progressively relaxes to lower levels of conductance, while displaying a large amount of current fluctuations. Since there is no plug, the return to lower levels of conductance may rather be due to occlusion of the pore lumen by the NTD and CTD, or partial collapse of the beta-barrel itself. We envision that the same processes might also occur in the WT channel, especially soon after displacement, since the dwell time at the dimeric current level is often extremely short. Unfortunately, as already mentioned previously (Mapingire et al., Citation2009), a mutant lacking both the PD and the globular domains expresses poorly, precluding any electrophysiological analysis to assess the behavior of the sole barrel. In comparison to the plugless PapC mutant, WT channels that contain a plug have a slower kinetics of return to the quiet level of activity similar to that observed prior to displacement; they often dwell at intermediate levels of conductance, and sometimes undergo a re-displacement event, as seen in . Since we find that new displacement events can be triggered again, it appears that plug re-entry does occur, perhaps on a slow time scale. Surprisingly, we observe that new displacements often require higher and higher voltages, as if the channels have become somewhat resistant to re-opening. This may be due to the channel adopting a desensitized state, as it is clear that the usher samples a variety of conformations with slow kinetics (see below).
Figure 3. Patch-clamp recording of the activity of the plugless PapC mutant. (A) The voltage protocol is depicted above the trace. As soon as the voltage is applied, the current jumps to the dimeric current level, but quickly relaxes to lower values. ‘C’ indicates the current level in the closed state of the usher, ‘M’ the monomeric level of conductance, and ‘D’ the dimeric level of conductance. (B) Amplitude histogram of the trace in panel A, shown in conductance to allow comparison with other amplitude histograms in Supplementary Figures (available online). The bin size is 0.003 nS.
Electrophysiological analysis of electrostatic network mutants
To determine whether specific electrostatic networks within the usher play a role in modulating plug displacement for pilus assembly, we investigated the D-quad, R-quad, and R-pairs mutants by patch-clamp electrophysiology. At voltages too low for triggering plug displacement, the mutants displayed an electrophysiological signature similar to that of WT. Upon application of a threshold voltage, all mutants were able to undergo plug displacement ( and ). Since there is some variability in this voltage between mutants (see below) and to some extent from patch to patch in the same mutant, the traces shown in and were obtained at different voltages. In stark contrast to the D-quad and R-quad mutants, the threshold voltage needed to trigger displacement in the R-pairs mutant was ∼40 mV lower than in the WT (). This suggests that disturbing the electrostatic interactions between these arginine pairs weakens the ability of the PD to gate the channel shut. In addition, while the WT activity typically returns to normal when low voltages are again applied after a plug displacement event, the R-pairs mutant usher maintains a very active state even when returned to low membrane potentials, as illustrated in (compare traces of ). All mutants, though, displayed the same relaxation behavior after displacement as seen in WT. Amplitude histograms show that the mutants sample a variety of kinetic states with similar conductances as the WT (Supplementary Figure 2, available online).
Figure 4. Plug displacement events in PapC mutants. Ten-second recordings of activity are shown for the indicated mutants. Since the average voltage threshold for plug displacement varies between the mutants (see ), the traces shown were obtained at different voltages: −140 mV for D-quad, −150 mV for R-quad, −110 mV for R single pair IN and R single pair OUT. For this reason, the traces are shown in conductance units (pS) to simplify the comparison between recordings. ‘C’ indicates the current level in the closed state of the usher, ‘M’ the monomeric level of conductance, and ‘D’ the dimeric level of conductance.
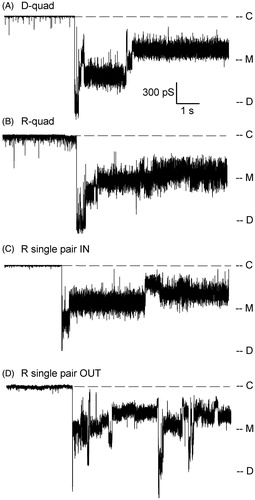
Figure 5. Electrophysiological analysis of the R-pairs PapC mutant. (A) Channel kinetics of the R-pairs mutant at −50 mV, with few short-live small openings before plug displacement. (B) In the same patch, applying a voltage of −100 mV triggered a plug displacement event. (C) Recording from the same patch at −50 mV after plug displacement. The R-pairs mutant channel typically remains active after plug displacement, even when the voltage is lowered back to values where the channel was relatively inactive before displacement. For all panels, the asterisk represents the beginning of the expanded view located below the trace, and ‘C’, ‘M’ and ‘D’ mark the current levels corresponding to the closed, open monomer and open dimer channels, respectively. The downward arrow in panel (A) points in the direction of channel openings.
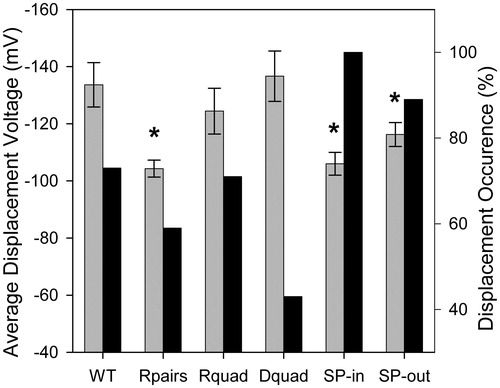
To quantify the effect of mutations on channel gating, we analyzed our experiments with respect to frequency of occurrence and voltage threshold for plug displacement (, ). Plug displacement was observed in 11 out of 15 experiments on WT channels, with an average displacement voltage of −134 mV. Although occurring much less frequently than in WT (three out of seven experiments), plug displacement required the same average voltage in the D-quad mutant than in WT. The R-quad mutant was similar to WT, whereas the most drastically affected mutant was the R-pairs mutant with an average threshold voltage for plug displacement of −104 mV. To determine whether this increased sensitivity originates from one of the pairs of arginines, we created mutants in a single pair, either at the inner surface of PapC (R single pair IN, ‘SP-in’: R256A - R332A), or at the outer surface of PapC (R single pair OUT, ‘SP-out’: R237A - R305A). These mutant ushers also exhibited plug displacement (). The threshold voltage of the mutant in the arginine pair closest to the periplasm (SP-in) was similar to that of the R-pairs mutant (−106 mV), while that of the external single R pair mutant (SP-out) was slightly less reduced relative to WT (−116 mV), but still significantly different. Both mutants had an increased occurrence of plug displacement, relative to WT. They also showed the typical current relaxation after plug displacement, and had a propensity to remain in an active form even at lower voltages, as seen for the R-pairs mutant (). Notably, the HA titer for the SP-in mutant was 0 (as in the R-pairs mutant), while it was as WT for the SP-out mutant. Therefore, it appears that the cellular and electrophysiological phenotypes of the R-pairs mutant are best recapitulated in the SP-in mutant, suggesting a prime role of the repulsive interaction between R256 and R332 in controlling the stability of the plug, and the ability of the usher to assemble a functional pilus.
Figure 6. Comparison of the displacement threshold voltages and occurrence rate between WT and mutants. The mutated residues in the R-pairs, R-quad and D-quad mutants are given in . SP-in is R256A - R332A, and SP-out is R237A - R305A. Gray and black bars represent the average displacement threshold voltage (left ordinate) and the % displacement occurrence (right ordinate), respectively. Note the negative values on the left ordinate as displacement never occurred at positive voltages. The (*) denotes statistical significance at the p = 0.05 level. Error bars are SEM. The numbers (N) of samples used for the average displacement threshold voltage are: 11 for WT, 7 for R-pairs, 9 for R-quad, 3 for D-quad, 5 for SP-in and 8 for SP-out. There are no error bars for the % occurrence, since this is not an average, but a value computed from all the experiments.
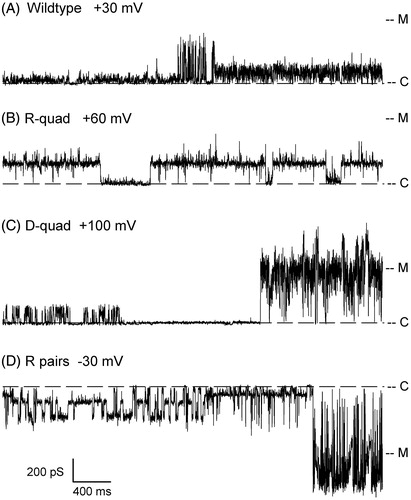
Dynamic plasticity of the usher and modal gating
We have observed that displacement of the plug is often followed by a period of activity characterized by a large amount of current transitions with distinctive kinetic signatures, open probabilities and conductance levels. This modal gating is frequent in the electrostatic network mutants investigated, but also apparent, on occasion, in WT channels. shows examples of gating modes from these four different types of proteins. The channels spontaneously switch between different gating modes, even during a sweep at a maintained membrane potential (see panels A, C and D for examples), illustrating that the protein is sampling a variety of conformational states. We attempted to categorize these behaviors in order to try to establish correlations with the nature of the channel, or voltage, or other parameter. Since the same gating modes may be seen in multiple variants, it was difficult to pinpoint any relationship between a gating mode and a mutant type. Rather, it appears that the mutations simply help the channel attain a level of dynamic activity after plug displacement, where this multiplicity of conformational states becomes more readily observable.
Figure 7. Modal gating of WT and electrostatic network mutants. Examples of gating modes observed in each PapC construct at the voltages indicated. To facilitate comparisons between panels, the traces are shown in conductance, rather than current. ‘C’ and ‘M’ denote the closed and monomeric current levels, respectively. Each trace is 4 sec-long. Switching between gating modes is observed in panels A, C and D.
Discussion
The outer membrane of Gram-negative bacteria acts as molecular sieve. Small molecules can freely pass through general diffusion porins, while larger ones are held at bay, either because they are harmful external agents, or necessary periplasmic components. However, this membrane also contains pore-forming proteins whose function is to translocate large substrates, such as folded polypeptides. To maintain the selective permeability barrier of the outer membrane, these large pores are often obstructed by constrictive elements, such as plugs, large extracellular loops, or accessory subunits. Examples can be found in TonB-dependent porins, such as BtuB or FepA (Nikaido, Citation2003), members of the Omp85 superfamily, such as FhaC and BamA (Guerin et al., Citation2014; Noinaj et al., Citation2015), the LptD-LptE complex for lipopolysaccharide insertion (Dong et al., Citation2014), and secretins (Tosi et al., Citation2014). The usher proteins are also large β-barrels where the channel is gated shut by a plug domain (Phan et al., Citation2011; Remaut et al., Citation2008). In many of these examples, the functionality of the protein itself relies on the conformational dynamics of the constricting element, which must relocate to allow the passage of substrates through the pore. Deciphering the molecular interactions that control the stabilization and movement of these constricting elements constitutes an essential step in understanding these complex molecular machines.
Two salt bridge networks were previously suggested to help localize the PD of PapC within the barrel lumen (Remaut et al., Citation2008). Two additional repulsive interactions that may play a role of in the stabilization of the PD within the barrel were also noted previously (Remaut et al., Citation2008). All of these networks link PD residues to specific amino acids on structural elements that are known to impact the open state of the channel, namely the α-helix and the β5–6 hairpin (Mapingire et al., Citation2009; Volkan et al., Citation2013). We previously demonstrated that deletion of the α-helix resulted in a PapC channel that has a tendency to undergo large opening transitions of size comparable to that of a plugless mutant (Mapingire et al., Citation2009). This was interpreted as spontaneous plug displacement events due to an unstable plug. This observation was recently supported by antibiotic sensitivity experiments, showing that the deletion of the α-helix or the β5–6 hairpin results in a significant increase in antibiotic sensitivity, presumably due to a leaky, occasionally unplugged, channel (Volkan et al., Citation2013). Increased antibiotic sensitivity was also observed in a single mutant with an alanine substitution at R305, a residue located on the plug domain and interacting with the E467 on the α-helix via a salt bridge. The R305A mutant also displayed an increased propensity at spontaneous large opening events in electrophysiological experiments. Alanine substitution at E467 also increased antibiotic sensitivity, and restoring the salt bridge with a double mutation that reversed the charges of both partners (R305E - E467R) returned the antibiotic sensitivity of the strain to WT levels. This observation supports the interpretation that the salt bridge is important. Here, we have extended these investigations by analyzing a quadruple mutant (R-quad) that includes alanine substitutions at R305 and E467, in addition to two other residues with which they may also interact. However, based on voltage threshold or displacement occurrence, we did not see a drastic effect on plug stabilization in this quadruple mutant relative to WT. Although we do not yet know the basis for this discrepancy, it should be kept in mind that the time frames of the two assays are different by orders of magnitude. Perhaps a slightly decreased plug stabilization (the R-quad mutant has a slightly lower threshold voltage) can translate into a noticeable effect on antibiotic permeability measured over several hours.
The R305 residue is also involved in a repulsive interaction with R237. Alanine substitutions at both these residues lead to a channel with a higher displacement occurrence than WT and a decreased threshold voltage. Therefore the pivotal role of R305 in plug stabilization may be related more to its involvement in this repulsive interaction than the salt bridge with E467. In addition, the repulsive interaction between the arginines located on the two P-linkers (R256 and R332) appears to play a major role in plug stabilization. We can imagine that it may act as a wedge keeping the two linkers apart and preventing the plug from exiting the barrel lumen. It is easily conceivable that the two linkers must indeed move relative to each other when the plug relocates towards the periplasmic side. This relative movement may be facilitated when the repulsive interaction between these residues is disrupted by the alanine substitutions. If electrostatic repulsion within these pairs of arginine is important for maintaining the plug in place, it is conceivable that raising the ionic strength of the solutions might reduce the electrostatic effect and essentially mimic the effect of alanine mutagenesis. However, in planar lipid bilayer experiments on the WT usher in 1 M KCl, the threshold voltage for plug displacement was −136 ± 12 mV (n = 5) and was essentially identical to the one obtained in 150 mM KCl. Upon examination of the PapC structure, the extracellular pair of arginine (R305-R237) is fairly buried within the bulk of the protein and not readily accessible to the solvent. Similarly, the pair found on the linkers (R256-R332), although located at the periplasmic surface of the barrel domain, is likely to be masked by the N- and C-globular domains. Therefore, we attribute the lack of effect of the ionic strength on the threshold voltage to the reduced accessibility of these pairs to the bulk solvent solution.
Other regions have also been identified as participants in the functionality of PapC. The quadruple mutations at one of the electrostatic networks investigated here (D-quad) yielded an usher with a voltage threshold similar to WT but that is less prone to plug displacement (; plug displacement occurrence is 43% relative to 73% in WT). Therefore, this network may play a role in plug release. The β12–13 loop, on the periplasmic face of the protein, comes in close contact with the displaced PD during translocation (Phan et al., Citation2011). The mutations of residues on this loop or on the 13th β-strand at the edge of the loop appear to yield channels less capable of opening than WT (Farabella et al., Citation2014; Volkan et al., Citation2013), suggesting that this region might be involved in maintaining the plug in position outside of the barrel pore once it has been displaced. The β12–13 loop could be conceived as a latch controlling channel gating. Recently, Farabella et al. (Citation2014) proposed that this latch might be regulated by an allosteric network of residues providing an intramolecular signaling pathway. With the use of a hybrid approach combining sequence coevolution analysis and molecular dynamics, critical residues were identified in four communities at the interface of the α-helix, β-hairpin, and plug domain. Pilus biogenesis, antibiotic sensitivity and electrophysiological phenotypes were analyzed in alanine substituted mutants. This analysis disclosed some residues that might have been missed as gating determinants by visual inspection only, in particular hydrophobic residues, such as V327 and F438, along with T331, which, once mutated into alanine, showed a greater propensity at plug displacement than WT. Interestingly, these residues are located on the P-linker 2 (V327, T331) or on the barrel wall opposite to P-linker 2 (F438). This observation reinforces the notion that the conformation of the P-linkers plays a determining role in plug dynamics. The alanine substitution of two other amino acids (K427 at the periplasmic surface, and S444 at the extracellular surface), though, led to a channel more reluctant to open than WT (Farabella et al., Citation2014).
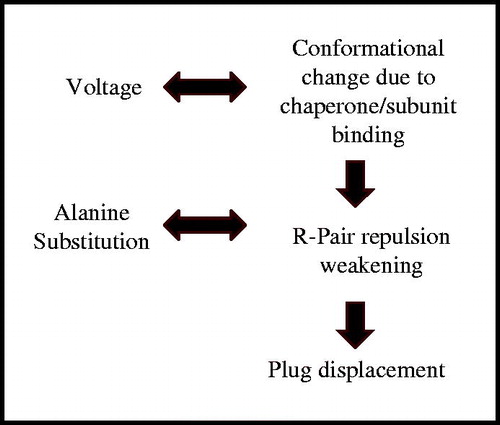
Taking these results together, we may envisage a pathway stretching from the periplasmic end to the extracellular surface of the protein that might propagate conformational changes initially triggered by the binding of the first chaperone-subunit complex to the usher NTD (Werneburg et al., Citation2015). This pathway would include the P-linkers, the β12–13 loop, the β-hairpin, the α-helix, and the plug domain, at the critical residues identified previously (Farabella et al., Citation2014). Conformational changes along this pathway might ultimately result in the relative movements of the arginine residues investigated here and the weakening of the repulsive force between them, essentially lessening the interactions that maintain the plug stably lodged into the barrel pore (). By introducing alanine mutations at the pairs of arginines involved in plug stabilization, we weakened the interactions even further, yielding channels that were even more sensitive to plug displacement. Charge displacements in response to the application of a transmembrane potential can lead to profound conformational changes with functional consequences, as seen for voltage-gated channels (Catterall, Citation2014). In the experiments reported here, high voltage was used as way to force the displacement of charged residues within the membrane electric field, i.e., as a mimic of the conformational changes brought about by chaperone-subunit binding in vivo (). We do not suggest that voltage is required for channel gating in vivo, as it is unlikely that the outer membrane maintains a membrane potential. In summary, we propose a model of usher gating whereby the binding of the first chaperone-subunit complex triggers conformational changes of the usher propagated by a previously identified allosteric network (Farabella et al., Citation2014) and leading to the weakening of the repulsion at the R256-R332 and R237-R305 pairs (). This weakened repulsion would represent a necessary step for destabilization of the PD within the barrel, leading to displacement and activation of the PapC usher for pilus biogenesis.
Figure 8. Summary of events leading to plug displacement. The in vivo sequence of events is depicted as a vertical workflow on the right hand side, while in vitro manipulations mimicking these steps are shown on the left hand side.
Conclusions
The PapC usher serves as an assembly and translocation platform for the biogenesis of P pili at the surface of uropathogenic E. coli. Our previous electrophysiological work demonstrated that ion conduction through PapC is severely restricted by the large plug domain present within the lumen of the channel, but that the protein has a highly dynamic behavior (Mapingire et al., Citation2009). Structural data reveal that the plug domain is displaced towards the periplasmic side during pilus assembly, thereby freeing up the pore for translocation of the folded pilus subunits through the outer membrane (Phan et al., Citation2011). The destabilization of the plug domain within the channel and ensuing displacement therefore represent critical steps in pilus biogenesis. Here we have demonstrated that specific mutations in PapC destabilize the plug domain and allow the protein to be more easily opened. Specifically, stabilization of the PapC plug domain is controlled by an electrostatic network of two arginine pairs, located close to the outer and inner surface of the plug domain. Although the involvement of repulsive interactions might seem counterintuitive in a stabilizing process, our results suggest that weakening of these interactions is a necessary step in the gating mechanism that opens the pore for pilus subunit transport. Together with other structure-function relationship studies (Farabella et al., Citation2014; Volkan et al., Citation2013), these observations provide mechanistic insights into PapC function and establish a basis for the design of therapeutic agents.
Supplementary material available online
Supplementary Table 1 and Supplementary Figures 1 and 2.
IMBC_1160450_Supplementary_Material.pdf
Download PDF (249.6 KB)Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by National Institutes of Health grants R01GM062987 (to D.G.T) and F30AI112252 (to G.T.W.).
Related Research Data
References
- Blomfield IC, Mcclain MS, Eisenstein BI. 1991. Type 1 fimbriae mutants of Escherichia coli K12: characterization of recognized afimbriate strains and construction of new fim deletion mutants. Mol Microbiol 5:1439–1445
- Busch A, Waksman G. 2012. Chaperone-usher pathways: diversity and pilus assembly mechanism. Philos Trans R Soc Lond B Biol Sci 367:1112–1122
- Catterall WA. 2014. Structure and function of voltage-gated sodium channels at atomic resolution. Exp Physiol 99:35–51
- Crepin S, Houle S, Charbonneau ME, Mourez M, Harel J, Dozois CM. 2012. Decreased expression of type 1 fimbriae by a pst mutant of uropathogenic Escherichia coli reduces urinary tract infection. Infect Immun 80:2802–2815
- Delcour AH. 2013. Electrophysiology of bacteria. Annu Rev Microbiol 67:179–197
- Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, et al. 2014. Structural basis for outer membrane lipopolysaccharide insertion. Nature 511:52–56
- Eidam O, Dworkowski FS, Glockshuber R, Grutter MG, Capitani G. 2008. Crystal structure of the ternary FimC-FimF(t)-FimD(N) complex indicates conserved pilus chaperone-subunit complex recognition by the usher FimD. FEBS Lett 582:651–655
- Farabella I, Pham T, Henderson NS, Geibel S, Phan G, Thanassi DG, et al. 2014. Allosteric signalling in the outer membrane translocation domain of PapC usher. eLife. Available from: http://dx.doi.org/10.7554/eLife.03532
- Ford B, Rego AT, Ragan TJ, Pinkner J, Dodson K, Driscoll PC, et al. 2010. Structural homology between the C-terminal domain of the PapC usher and its plug. J Bacteriol 192:1824–1831
- Fronzes R, Remaut H, Waksman G. 2008. Architectures and biogenesis of non-flagellar protein appendages in Gram-negative bacteria. EMBO J 27:2271–2280
- Geibel S, Procko E, Hultgren SJ, Baker D, Waksman G. 2013. Structural and energetic basis of folded-protein transport by the FimD usher. Nature 496:243–246
- Geibel S, Waksman G. 2014. The molecular dissection of the chaperone-usher pathway. Biochim Biophys Acta 1843:1559–1567
- Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA 87:4645–4649
- Guerin J, Baud C, Touati N, Saint N, Willery E, Locht C, et al. 2014. Conformational dynamics of protein transporter FhaC: large-scale motions of plug helix. Mol Microbiol 92:1164–1176
- Henderson NS, Ng TW, Talukder I, Thanassi DG. 2011. Function of the usher N-terminus in catalysing pilus assembly. Mol Microbiol 79:954–967
- Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–28
- Lee D, Lee J, Seok C. 2013. What stabilizes close arginine pairing in proteins? Phys Chem Chem Phys 15:5844–5853
- Li H, Qian L, Chen Z, Thibault D, Liu G, Liu T, Thanassi DG. 2004. The outer membrane usher forms a twin-pore secretion complex. J Mol Biol 344:1397–1407
- Li H, Thanassi DG. 2009. Use of a combined cryo-EM and X-ray crystallography approach to reveal molecular details of bacterial pilus assembly by the chaperone/usher pathway. Curr Opin Microbiol 12:326–332
- Lund B, Lindberg F, Marklund BI, Normark S. 1987. The PapG protein is the alpha-D-galactopyranosyl-(1—-4)-beta-D-galactopyranose-binding adhesin of uropathogenic Escherichia coli. Proc Natl Acad Sci USA 84:5898–5902
- Mapingire OS, Henderson NS, Duret G, Thanassi DG, Delcour AH. 2009. Modulating effects of the plug, helix, and N- and C-terminal domains on channel properties of the PapC usher. J Biol Chem 284:36324–36333
- Nakamura K, Mizushima S. 1976. Effects of heating in dodecyl sulfate solution on the conformation and electrophoretic mobility of isolated major outer membrane proteins from Escherichia coli K-12. J Biochem 80:1411–1422
- Ng TW, Akman L, Osisami M, Thanassi DG. 2004. The usher N terminus is the initial targeting site for chaperone-subunit complexes and participates in subsequent pilus biogenesis events. J Bacteriol 186:5321–5331
- Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656
- Nishiyama M, Horst R, Eidam O, Herrmann T, Ignatov O, Vetsch M, et al. 2005. Structural basis of chaperone-subunit complex recognition by the type 1 pilus assembly platform FimD. EMBO J 24:2075–2086
- Nishiyama M, Ishikawa T, Rechsteiner H, Glockshuber R. 2008. Reconstitution of pilus assembly reveals a bacterial outer membrane catalyst. Science 320:376–379
- Noinaj N, Rollauer SE, Buchanan SK. 2015. The beta-barrel membrane protein insertase machinery from Gram-negative bacteria. Curr Opin Struct Biol 31:35–42
- Phan G, Remaut H, Wang T, Allen WJ, Pirker KF, Lebedev A, et al. 2011. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 474:49–53
- Prilipov A, Phale PS, Van Gelder P, Rosenbusch JP, Koebnik R. 1998. Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol Lett 163:65–72
- Remaut H, Tang C, Henderson NS, Pinkner JS, Wang T, Hultgren SJ, et al. 2008. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell 133:640–652
- Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, et al. 1994. The Gal(alpha 1-4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc Natl Acad Sci USA 91:11889–11893
- Sauer FG, Futterer K, Pinkner JS, Dodson KW, Hultgren SJ, Waksman G. 1999. Structural basis of chaperone function and pilus biogenesis. Science 285:1058–1061
- Simonet VC, Basle A, Klose KE, Delcour AH. 2003. The Vibrio cholerae porins OmpU and OmpT have distinct channel properties. J Biol Chem 278:17539–17545
- So SS, Thanassi DG. 2006. Analysis of the requirements for pilus biogenesis at the outer membrane usher and the function of the usher C-terminus. Mol Microbiol 60:364–375
- Thanassi DG, Bliska JB, Christie PJ. 2012. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol Rev 36:1046–1082
- Tosi T, Estrozi LF, Job V, Guilvout I, Pugsley AP, Schoehn G, Dessen A. 2014. Structural similarity of secretins from type II and type III secretion systems. Structure 22:1348–1355
- Volkan E, Kalas V, Pinkner JS, Dodson KW, Henderson NS, Pham T, et al. 2013. Molecular basis of usher pore gating in Escherichia coli pilus biogenesis. Proc Natl Acad Sci USA 110:20741–20746
- Vondrasek J, Mason PE, Heyda J, Collins KD, Jungwirth P. 2009. The molecular origin of like-charge arginine-arginine pairing in water. J Phys Chem B 113:9041–9045
- Werneburg GT, Henderson NS, Portnoy EB, Sarowar S, Hultgren SJ, Li H, Thanassi DG. 2015. The pilus usher controls protein interactions via domain masking and is functional as an oligomer. Nat Struct Mol Biol 22:740–746