Abstract
Obesity is fast becoming the scourge of our time. It is one of the biggest causes of death and disease in the industrialized world, and affects as many as 32% of adults and 17% of children in the USA, considered one of the world's fattest nations. It can also cost countries billions of dollars per annum in direct and indirect care, latest estimates putting the USA bill for obesity-related costs at $147 billion in 2008. It is becoming clear that the pathophysiology of obesity is vastly more complicated than the simple equation of energy in minus energy out. A combination of genetics, sex, perinatal environment and life-style factors can influence diet and energy metabolism. In this regard, psychological stress can have significant long-term impact upon the propensity to gain and maintain weight. In this review, we will discuss the ability of psychological stress and ultimately glucocorticoids (GCs) to alter appetite regulation and metabolism. We will specifically focus on (i) GC regulation of appetite and adiposity, (ii) the apparent sexual dimorphism in stress effects on obesity and (iii) the ability of early life stress to programme obesity in the long term.
Glucocorticoid regulation of appetite and adiposity
Psychological stress is a significant risk factor for obesity
Severe psychological trauma can clearly contribute significantly to obesity in later life. Studies have shown that post-traumatic stress disorder, a debilitating anxiety disorder that develops after exposure to a single or multiple extremely traumatic events, is a significant predictor of obesity (Vieweg et al. Citation2007; Kozaric-Kovacic et al. Citation2009; Perkonigg et al. Citation2009). Similarly, exposure to war-related events during childhood has been associated with a higher body mass index (BMI) 10 years later (Llabre and Hadi Citation2009). More mild psychological stress is also closely associated with excess weight gain. For instance, female macaques exposed to the chronically stressful social subordination or social reorganization have increased visceral fat deposition and adverse metabolic characteristics such as hyperglycaemia (Shively and Clarkson Citation1988; Jayo et al. Citation1993; Shively Citation1998; Shively et al. Citation2009). Numerous studies in humans have established a link between an environment of stress and excess body weight. Social status (defined by employment grade) is inversely related to central adiposity and the metabolic syndrome in men and women (Brunner et al. Citation1997). Perceived psychosocial stress in multiple domains, such as job-related demands and difficulty paying bills, has also been linked with weight gain over an extended period in adult men and women (Block et al. Citation2009; Fowler-Brown et al. Citation2009). In addition to, job stress being associated with obesity, it has also been associated with negative eating behaviours such as eating to satiety and to curb irritability (Nishitani et al. Citation2009). Psychological stress also contributes to obesity in children. Children from families reporting high levels of stress across a number of domains are significantly more likely to be obese than those from “low-stress” families (Koch et al. Citation2008; Moens et al. Citation2009), with serious events such as divorce, parental unemployment or death of a family member having particular impact.
While a background of psychological stress can confer a propensity to develop obesity, the condition itself is linked to exacerbated responses to stress. For instance, the cortisol response to public speaking stress is enhanced in obese women, as is heart rate and diastolic blood pressure (Benson et al. Citation2009). Obese people are also more likely to suffer from stress, anxiety and depression (Doyle et al. Citation2007; Scott et al. Citation2008; Abiles et al. Citation2010). Whether this phenomenon is responsible for or symptomatic of obesity cannot be deduced from these studies. However, a more direct link between psychological stress, a hyperactive hypothalamic–pituitary–adrenal (HPA) axis and obesity in humans has recently been established. Thus, women who developed obesity following a stressful event were found to have gained weight significantly faster than those who developed obesity without a prior stressful event, and this was associated with higher basal cortisol levels (Vicennati et al. Citation2009). It is, therefore, emerging that dysfunctional regulation of the stress response, particularly of the HPA axis, after severe or prolonged stress is significantly involved in the pathogenesis of obesity.
Glucocorticoid regulation of appetite
The HPA axis is the key endocrine axis responsible for mediating the body's response to stress. Other components of the body's response to stress such as sympathomedullary activation undoubtedly also influence obesity, but are beyond the scope of this review. Briefly, when an organism encounters a stressor, medial parvocellular cells in the paraventricular nucleus of the hypothalamus (PVN) are activated, leading to the release of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) at the median eminence into the hypothalamo-hypophysial portal blood vessel system. Adrenocorticotropic hormone is then released from the anterior pituitary into the systemic circulation, and acts at the adrenal cortex to stimulate glucocorticoid (GC: i.e. cortisol in humans, corticosterone in rodents) synthesis and release. GCs have a number of functions to assist in combating the stressor, including to feed back and prevent further HPA axis activation. This axis has been reviewed extensively elsewhere (Sapolsky et al. Citation2000; Papadimitriou and Priftis Citation2009). There are several mechanisms by which dysregulation of the HPA axis and of GC function may affect energy balance and potentially contribute to obesity.
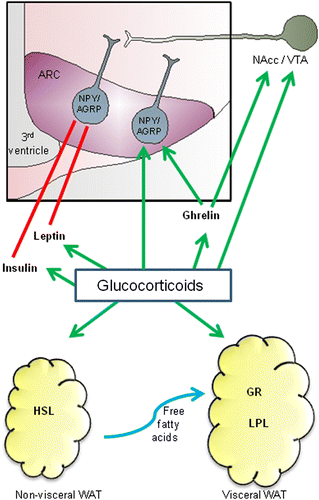
GCs are, for instance, important in appetite regulation (). Acutely after stress, appetite is suppressed. This anorexia occurs centrally via a CRH-mediated mechanism, whereby CRH (Heinrichs and Richard Citation1999; Richard et al. Citation2002) and other CRH-like molecules such as urocortin (Weninger et al. Citation1999; Richard et al. Citation2002) act in the brain to suppress appetite, probably via an inhibition of neuropeptide Y (NPY)-stimulated food intake (Heinrichs et al. Citation1993; Currie Citation2003). This CRH-induced anorexia is likely to involve a number of brain regions including the PVN, perifornical and ventromedial regions of the hypothalamus, lateral septum and parabrachial nucleus (Richard et al. Citation2002), but recent evidence also indicates an important role for the indirect action of CRH on the dorsal portion of the anterior bed nucleus of the stria terminalis (Ciccocioppo et al. Citation2003). Although stress may acutely suppress appetite in this way, GCs are released in the hours to days following a stressful event and these stimulate feeding (Santana et al. Citation1995; Dallman et al. Citation2004). Indeed, George et al. (Citation2010) were recently able to show that elevated cortisol concentrations are correlated with food intake as little as 1 h after a peripheral injection of CRH in humans, and that the amount of food consumed is directly related to the size of the peak cortisol response (George et al. Citation2010).
Figure 1. An overview of the concepts discussed for GC regulation of appetite and adiposity. GCs stimulate food intake via direct action on orexigenic NPY/AGRP neurons in the ARC, as well by stimulating nucleus accumbens (NAcc)/VTA pathways that reinforce the rewarding nature of food. GC may also stimulate food intake by enhancing the appetite- and reward-stimulatory effects of ghrelin. In addition, GC can prevent the actions of satiety hormones leptin and insulin; reducing the sensitivity of the brain to these hormones and contributing to leptin and insulin resistance. GC also contributes to enhancing visceral fat WAT. They activate HSL, enhancing lipolysis and LPL, promoting fat storage. Because there is a greater density of GRs in visceral fat, as well as greater LPL activity, fat storage may be preferentially promoted in this tissue. See text for details.
In an acutely stressful situation, this is probably a highly adaptive phenomenon. The stimulation of feeding would serve to replace lost energy and to facilitate a “stocking up” in anticipation of the next stressor. However, if the stressor continues too long or there are multiple sequential stressors, GC levels can become chronically elevated, leading in humans to a chronically stimulated appetite, increased feeding and consequently to obesity (De Vriendt et al. Citation2009).
GCs stimulate food intake via a series of complex interactions on various appetite-regulating targets. They stimulate the actions of orexigenic peptides NPY and agouti-related peptide (AGRP) (Savontaus et al. Citation2002; Konno et al. Citation2008), up-regulating gene expression of NPY and AGRP in the arcuate nucleus (ARC) by increasing AMP-activated protein kinase signalling (Shimizu et al. Citation2008). GCs also influence the function of anorexigenic leptin. Although GCs stimulate leptin release from adipose tissue, which would normally lead to appetite suppression, they also reduce the sensitivity of the brain to leptin, contributing to leptin resistance (Zakrzewska et al. Citation1997, Citation1999; Jequier Citation2002).
In addition to leptin, GCs also stimulate insulin secretion from the pancreas (Strack et al. Citation1995), which has complex effects on appetite. Insulin usually acts at the hypothalamus to reduce food intake and at the ventral tegmental area (VTA) to reduce the dopaminergic neuron-mediated rewarding nature of food (Figlewicz et al. Citation2008). However, when food choice is available, it enhances the preference for “comfort foods”, particularly those high in fat and sucrose (la Fleur et al. Citation2004; Warne et al. Citation2006, Citation2009). In addition, chronically elevated GC concentrations can contribute to insulin resistance, reducing its ability to inhibit feeding-stimulatory pathways in the brain, as is the case with leptin (Asensio et al. Citation2004).
An emerging, but still poorly elucidated mechanism by which GC may further influence appetite regulation during stress is via ghrelin. Ghrelin is a gastrointestinal peptide usually associated with feeding regulation, acting to signal initiation of a meal (Hosoda et al. Citation2006). Circulating levels of ghrelin are increased during stress (Kristenssson et al. Citation2006) and activate CRH and AVP release to potentiate the stress response, probably doing so either directly by acting at the PVN or indirectly by activating brainstem noradrenergic inputs to the PVN (Cummings Citation2006; Kawakami et al. Citation2008). It is, therefore, conceivable that chronic or severe stress resulting in prolonged elevated GC secretion could lead to chronically up-regulated ghrelin signalling, culminating in increased food intake.
Stress and GCs tend to stimulate appetite specifically for high-energy foods, with insulin having a key role in this regard (la Fleur et al. Citation2004; Warne et al. Citation2006, Citation2009; Dallman Citation2010). It has been established that chronically stressed animals prefer calorically dense foods and that these can actually ameliorate HPA axis responses to further stress (Pecoraro et al. Citation2004; Foster et al. Citation2009). Thus, rats exposed to chronic restraint stress voluntarily ingested more “comfort food” (lard and sucrose) than non-restrained rats, and HPA axis responses to the restraint were reduced in rats that had access to these high-calorie foods (Pecoraro et al. Citation2004). Given the strongly rewarding nature of this cycle, it is not surprising that this preference for highly palatable foods during stress is mediated through some of the same pathways that mediate responses to drugs of addiction (Nieuwenhuizen and Rutters Citation2008; Coccurello et al. Citation2009). Dopaminergic, opioid and glutamatergic transmission within the nucleus accumbens, for instance, appear to be crucial for mediating reward-associated information, whether it be regarding drugs (Di Chiara and Imperato Citation1988; Koob and Le Moal Citation2001; Ito et al. Citation2004), sex (Balfour et al. Citation2004) or palatable food (Berridge Citation2009). This reward circuitry has long been known to be activated by eating (Bassareo and Di Chiara Citation1997, Citation1999) and, interestingly, the propensity to ingest highly palatable food has been correlated with preference for drugs of abuse (Gosnell Citation2000). For instance, ghrelin, which usually serves to stimulate food intake (Hosoda et al. Citation2006) and subserves the rewarding nature of food (Egecioglu et al. Citation2010), is also essential for the rewarding effects of alcohol and psychostimulant drugs (Jerlhag et al. Citation2009, Citation2010).
Glucocorticoid regulation of adiposity
Stress can clearly contribute to the propensity to obesity via GC-mediated up-regulation of appetite, particularly for fatty foods. However, GCs also play a role in the regulation of lipid homeostasis (fat mobilization from adipose tissue and fat deposition and storage; ).
Excess GC has been particularly associated, in primate models (Shively and Clarkson Citation1988; Jayo et al. Citation1993; Shively Citation1998; Shively et al. Citation2009) and in humans, with visceral fat accumulation (Marin et al. Citation1992b; Rosmond et al. Citation1998; Epel et al. Citation2000), a phenotype that is strongly linked to an increased risk for cardiovascular disease (Grassi et al. Citation2004; Hamdy et al. Citation2006; Mathieu et al. Citation2008). GC receptors (GRs) are present in visceral adipose tissue in greater density than in other adipose depots (Rebuffe-Scrive et al. Citation1990) and may serve to relatively enhance the effects of GC specifically in this tissue (Bjorntorp Citation2001). Additionally, type 1 11β-hydroxysteroid dehydrogenase (11β-HSD1), an enzyme which generates active GC from inactive GC metabolites within tissues, has been shown in mice to be linked to visceral adiposity, with over-expression of the 11β-HSD1 gene leading specifically to visceral fat accumulation (Masuzaki et al. Citation2001). Conversely, transgenic mice expressing specifically in adipocytes type 2 11β-HSD (11β-HSD2), which inactivates GC, protects against this phenotype (Kershaw et al. Citation2005). In humans, increased visceral fat 11β-HSD1 oxoreductase activity is also associated with increased accumulation in this depot, as well as adipocyte hypertrophy, increased lipolysis (the release of non-esterified fatty acids from adipocytes) and increased lipoprotein lipase (LPL) activity in this tissue (Veilleux et al. Citation2009). It appears that elevated 11β-HSD1 may be a cause rather than a consequence of obesity, as inhibiting the enzyme can improve metabolic parameters and reduce weight (Tiwari Citation2010). Furthermore, disruption of 11β-HSD1 leads to protection from high-fat diet-associated weight gain and a fat distribution profile away from the visceral depot (Morton et al. Citation2004). These studies clearly demonstrate a relationship between GC activity and adiposity; however, the mechanisms underpinning this relationship are not clearly understood.
The action of GC on fatty acid metabolism and storage is complex and GCs have a variety of direct and permissive effects (Macfarlane et al. Citation2008; Xu et al. Citation2009). It is generally seen that GC acutely (hours) enhance lipolysis. That is, short-term infusion of GC in vivo stimulates the release of non-esterified fatty acids from adipocytes (Divertie et al. Citation1991) through the activation of hormone-sensitive lipase (HSL) (Slavin et al. Citation1994), a key enzyme responsible for enhancing fatty acid mobilization (). This increase in circulating free fatty acids may restrict glucose utilization and encourage insulin resistance (Arner Citation2002). GCs have also been shown to enhance adipose LPL activity, which should promote fat storage (Bjorntorp Citation1996, Citation2001). Thus, it appears that GC may enhance lipid metabolism through its effects on both the turnover and uptake of fatty acids in adipose tissue. Since LPL activity is greater in visceral adipose tissue than in other adipose depots (Marin et al. Citation1992a), GCs may, in this way, contribute to a redistribution of fat such that it accumulates preferentially in visceral depots (). Insulin appears to be essential for this process as it is able to markedly potentiate the GC affect on LPL (Ashby and Robinson Citation1980).
GCs also play a critical role in the differentiation of pre-adipocytes into mature adipocytes (Hauner et al. Citation1987; Tomlinson and Stewart Citation2002), a role which is again insulin related (Hauner et al. Citation1989; Gregoire et al. Citation1992; Suryawan et al. Citation1997), potentially further accentuating the accumulation of fat. They are also known to up-regulate the NPY Y2 receptor in abdominal fat, leading to stimulation of proliferation and differentiation of adipocytes, again culminating in enhanced fat accumulation (Kuo et al. Citation2007, Citation2008). Although the distinct molecular mechanisms by which GCs may affect adiposity remain to be determined, it is clear that active GCs are strong modulators of adipose tissue metabolism and distribution in rodents and humans, contributing to enhanced fat deposition particularly in the high-risk visceral adipose depot.
Sexual dimorphism in stress effects on obesity
Sexual dimorphism in stress effects on white adipose tissue
Chronic stress and the increased GC level that results can thus predispose an individual to obesity or exacerbate an already obese phenotype. However, there are indications that there is some sexual dimorphism in these effects.
Obesity affects men and women very differently. Women are more likely to develop obesity than men (Lovejoy and Sainsbury Citation2009), and are more likely to develop metabolic syndrome if they do become obese (Razzouk and Muntner Citation2009). They are also less likely to lose weight with calorie restriction or exercise regimes than men (Meijer et al. Citation1991; Westerterp and Goran Citation1997; Sartorio et al. Citation2005). Men, by contrast, have a higher risk of accumulating fat in the visceral depot, leaving them more vulnerable to associated complications such as cardiovascular disease (Bjorntorp Citation1996).
Several studies have suggested that women may be more sensitive to the obesity-promoting effects of psychological stress. For instance, women who are moderately overweight (BMI = 25.0–29.9) are more likely than normal weight women to report having experienced stressful life events. In men, obesity and extreme obesity are associated with more stressful life events, but a moderately overweight phenotype is not (Barry et al. Citation2008). Similarly, a 13-year follow-up study of black American adults revealed high levels of perceived stress is correlated with greater percentage increases in BMI in women, but not in men (Fowler-Brown et al. Citation2009). Our own studies in rats have suggested that females, but not males, that became obese due to overfeeding as neonates have enhanced HPA axis responses to psychological stress (Spencer and Tilbrook Citation2009).
BMI may, however, be a less useful predictor of the health outcomes associated with obesity than adipose distribution. Obese men and post-menopausal women have a significantly greater risk of developing obesity-associated diseases such as type II diabetes mellitus and cardiovascular disease than pre-menopausal women owing to a profile of enhanced visceral fat (Bjorntorp Citation1996). This profile in particular may be associated with differential effects of GC at the adipose tissue level (Tomlinson et al. Citation2004).
As mentioned, GCs may contribute to a predominantly visceral fat distribution. In women, lipolysis is stimulated more effectively in subcutaneous than in visceral adipocytes, whereas there are no differences between the depots in men. The synthetic GC, dexamethasone, is also able to increase the ability of a synthetic cAMP analogue to stimulate lipolysis in subcutaneous adipose tissue in women, with a similar tendency in visceral adipocytes, but not in men (Lundgren et al. Citation2008). Conceivably, the major action of GC in females is at subcutaneous rather than visceral fat to stimulate lipolysis, whereas in men GC would act at subcutaneous and visceral fat equally and promote visceral fat distribution via greater activation of LPL in this tissue.
A key player in this adipose depot specific action of GC is also 11β-HSD1. As mentioned, higher 11β-HSD1 activity in visceral vs. subcutaneous adipose tissue is associated with preferential visceral fat accumulation and associated metabolic alterations (Veilleux et al. Citation2009). Rat and human studies demonstrate significantly higher activity of 11β-HSD1 in males compared with females (Albiston et al. Citation1995; Weaver et al. Citation1998; Toogood et al. Citation2000), which may contribute to increased visceral adiposity in males in the face of chronic stress.
Sexual dimorphism in stress effects on brown adipose tissue
In addition to the action of GC at white adipose tissue (WAT), other sexually dimorphic peripheral effects of GC may contribute to sexually dimorphic effects of stress on obesity, for instance at the level of brown adipose tissue (BAT). BAT is responsible for dissipating energy as heat primarily via an uncoupling protein (UCP)1-mediated uncoupling of oxidative phosphorylation (Cannon and Nedergaard Citation2004). Impairments in BAT function can, therefore, result in reduced thermogenesis and thus increased energy storage leading to obesity. In this regard, BAT dysfunction is certainly a consistent factor across many models of obesity. Ablation of BAT will result in obesity even in the absence of hyperphagia (Lowell et al. Citation1993) and various genetic models of obesity, such as the ob/ob and db/db mice, are also found to have reduced levels of BAT thermogenesis (Commins et al. Citation1999; Masaki et al. Citation2000).
There is some evidence that BAT functions differently in males and females. In rodents, basal BAT thermogenesis is greater in ad libitum fed females than in males in a thermoneutral environment (Justo et al. Citation2005; Valle et al. Citation2007). Female rats are also capable of deactivating BAT thermogenesis to a greater degree than males under calorie-restricted conditions and this is associated with resistance to the loss of lean mass (Valle et al. Citation2005). In addition, we have recently found that females made obese by overfeeding during the neonatal period maintain an obese phenotype due to a down-regulation of BAT function, whereas males show no such alterations in BAT activity (Stefanidis, Tilbrook and Spencer unpublished observations, 2009–2010).
Until recently, it was thought that adult humans did not possess substantial amounts of active BAT. This dogma has now been dispelled by several conclusive high-profile studies revealing that BAT depots exist in the neck anterior to the thorax, particularly the cervical–supraclavicular region (Nedergaard et al. Citation2007; Cypess et al. Citation2009; van Marken Lichtenbelt et al. Citation2009; Virtanen et al. Citation2009; Zingaretti et al. Citation2009). It is noteworthy that these studies have revealed differences in the frequency with which men and women present with functionally active BAT—defined regions of active BAT being present more frequently in women than men (Cypess et al. Citation2009). These findings lead to the suggestion that the role of BAT in human obesity is also sexually dimorphic, or at least presents a sexually dimorphic target for therapeutic intervention.
With respect to the effects of stress on this tissue, GCs are among the strongest down-regulators of UCP1 expression in BAT (Arvaniti et al. Citation1998), contributing to a reduction in thermogenesis and a conservation of energy that could, in a chronic setting, promote obesity. The greater predominance of BAT in women and the sexually dimorphic sensitivity of BAT to environmental manipulations suggest that stress may also influence the development and maintenance of obesity in a sexually dimorphic manner via its effects at this tissue.
The role of sex steroids in obesity and the impact of stress
Obesity may also occur by different mechanisms in males and females due to the differential involvement of sex steroids. Sex steroid hormones, sexually dimorphic after puberty, act centrally to regulate feeding as well as peripherally to influence adipose tissue metabolism, and their actions may be stress sensitive. Receptors for oestrogen, progesterone and androgens are all present in WAT and BAT and the sex steroids can act as transcriptional regulators of various metabolic genes. For instance, oestrogen, the predominant sex steroid in females, can exert effects on LPL to alter lipid accumulation, playing an essential role in fat distribution (Shi et al. Citation2009) and leading to greater inguinal fat deposition in females (Bjorntorp Citation1996; Pedersen et al. Citation2004). Thus, ovariectomy in rodents increases food intake and promotes weight gain, and replacing oestradiol is sufficient to normalize this (Asarian and Geary Citation2002). Similarly, menopause in human females, leading to a reduction in oestrogen levels, is also associated with increased weight gain and a shift towards visceral fat deposition (Svendsen et al. Citation1995; Tchernof et al. Citation2000). Levels of progesterone and testosterone are also sexually dimorphic and may contribute to variation in energy balance (Lobo et al. Citation1993; Mayes and Watson Citation2004; Saad and Gooren Citation2009). For instance, testosterone also inhibits LPL activity and adipogenesis (De Pergola Citation2000; Blouin et al. Citation2010), and hypotestosteronaemia in human males has been associated with increased adiposity (Mah and Wittert Citation2010). Sexual dimorphism in levels of sex hormones or their receptors may also contribute to sexual dimorphism in BAT thermogenic capacity. Females have lower levels of BAT androgen receptors and oestrogen receptor α than males, both of which are involved in promoting thermogenesis (Rodriguez-Cuenca et al. Citation2007). These differences could potentially leave females more vulnerable to changes in concentrations of the ligand.
Stress and chronically elevated GC levels can have pronounced effects on androgen activity. In male rats, the circadian rise in corticosterone concentrations are associated with a dip in testosterone levels, and the normal bimodal circadian pattern of testosterone release can be abolished with the administration of synthetic GC (Waite et al. Citation2009). Thus, there is an antagonistic relationship between the activation of the HPA axis and that of the HPA axis, with chronic stress-suppressing sex hormone secretion and interfering with reproductive function (Retana-Marquez et al. Citation2003; Kinsey-Jones et al. Citation2009). The hypotestosteronaemia that results from an overactive HPA axis or from chronic stress in males may contribute in turn to increased adiposity and an obese phenotype (CitationMah and Wittert 2009), as may a chronic reduction in circulating oestrogen levels in females with HPA axis activation (Kinsey-Jones et al. Citation2009).
Stress, obesity and neonatal programming
Prenatal stress programmes a predisposition to obesity
Foetal and neonatal environmental risk factors are increasingly recognized as relevant determinants of adult physiology and may contribute to an obese phenotype in adulthood. It is likely that stress, at critical windows of development, is one of these risk factors.
Adaptations during pregnancy ensure that the foetus is relatively protected against increases in GC. Elevated levels of progesterone and its metabolite, allopregnanolone, in pregnancy ensure that there is an enhancement of the allopregnanolone-mediated inhibition of the noradrenergic input to the PVN from the nucleus of the solitary tract (Brunton et al. Citation2005, Citation2009; CitationBrunton and Russell 2010). Thus, pregnant females respond to stressors with less of an increase in GC than do non-pregnant females (Brunton et al. Citation2005, Citation2009; Slattery and Neumann Citation2008). Placental 11β-HSD2, responsible for deactivating GC, also plays a crucial role in ameliorating the impact of maternal GC (Lucassen et al. Citation2009). However, in cases of severe or chronic maternal stress, excess GC may still affect the foetus.
Studies in humans and in animal models have revealed myriad long-term effects on the offspring of maternal exposure to stress or GC during pregnancy. Stressful events during pregnancy can impact upon foetal brain development leading to alterations in HPA axis (Henry et al. Citation1994; Rossi-George et al. Citation2009), anxiety behaviours (Vallee et al. Citation1997), learning and memory (Lordi et al. Citation1997; Entringer et al. Citation2009), sensitivity to drug abuse (Morley-Fletcher et al. Citation2004; Thomas et al. Citation2009) and, crucially, obesity. For instance, maternal bereavement immediately before or during pregnancy is associated with a significantly increased risk of excess weight gain in childhood (Li et al. Citation2010).
One major factor contributing to the aetiology of our current obesity epidemic may, therefore, be excessive exposure to stress or GC at specific time points during prenatal or post-natal development. Prenatal (gestational days 8, 10 and 12) exposure to dexamethasone (a synthetic GC), or to the physical stressor (see (Dayas et al. Citation2001; Keay and Bandler Citation2001) for discussion on physical vs. psychological stress) of exposure to the cytokines interleukin 6 or tumour necrosis factor α, was seen to increase adiposity throughout life by up to 40% (Dahlgren et al. Citation2001). Such effects appear to be somewhat gender specific. Thus, while dexamethasone and these cytokines affected offspring adiposity in both males and females (Dahlgren et al. Citation2001), doses of lipopolysaccharide (LPS; 0.79 mg/kg) at the same gestational ages led to adiposity and insulin resistance in male but not female offspring (Nilsson et al. Citation2001).
However, prenatal stress alone may not necessarily be sufficient to induce obesity in the offspring (D'Mello and Liu Citation2006; Baker et al. Citation2008), but may still confer an increased susceptibility to obesity (Tamashiro et al. Citation2009). For instance, the offspring of rats exposed to chronic variable stress during the third week of gestation do not become obese if weaned onto a normal rat-chow diet. However, obesity and impaired glucose tolerance in adulthood are manifested if these offspring are weaned onto high-fat chow (Tamashiro et al. Citation2009). This susceptibility may be related, in part, to prenatal stress causing intrauterine growth restriction (reflected in lower birth weights (Lesage et al. Citation2004)) which is then compensated for by catch-up growth if sufficient high-energy food is available after birth (Hales and Barker Citation1992; Gluckman and Hanson Citation2004).
Post-natal events also influence development, and post-natal stress can also lead to a predisposition to obesity in adulthood. Early psychological trauma in humans, ranging from sexual abuse to the death of a mother or parental stress, is a significant risk factor for the development of obesity in later life (Koch et al. Citation2008; D'Argenio et al. Citation2009). In rodents, the effects of post-natal stress are less clear. It appears that exposure to stress in the early post-natal period may not directly lead to obesity but may confer an enhanced susceptibility. In rodents, separation from the mother is a significant psychological stress to the neonate and has long-lasting physiological effects, including potentially enhancing susceptibility to changes in energy balance and metabolism. Thus, neither separation from the mother as a neonate nor social isolation post weaning alone significantly affect body weight, but social isolation in rats that had previously undergone maternal separation leads to significant, long-lasting increases in food intake and weight gain (Ryu et al. Citation2009).
An important point highlighted by several of these investigations is that timing is critical. Chronic exposure to dexamethasone in early gestation in the common marmoset does not adversely affect the animal, while the same type of challenge late in gestation leads to early indices of metabolic syndrome such as high fasting plasma glucose and triglyceride levels (Nyirenda et al. Citation2009). Restraint stress experienced in rats in mid-late gestation produces long-term changes in the body weights of offspring of stress-sensitive dams, whereas early gestation restraint stress does not (Mueller and Bale Citation2006). Similarly, the physical stressor of an immune challenge with LPS experienced at either post-natal days 3, 7 or 14 does not affect long-term growth or adiposity in the pair-housed rat (Spencer et al. Citation2007), whereas a similar challenge presented at day 10 does appear to mildly increase adult body weight (Iwasa et al. Citation2010). Interestingly, it appears that the programming effects of stress may not be confined to the perinatal period. Chronic social stress has also been shown to lead to long-term alterations in HPA axis function and fat distribution if it occurs during adolescence (Schmidt et al. Citation2009).
Equating developmental stages between species is fraught with controversy. However, it is generally considered that late gestation and the early post-natal period in the rodent are roughly equivalent to the third trimester of pregnancy in the human, particularly with regard to metabolic systems (Grove et al. Citation2005). Thus, it appears that, for humans, stress encountered during the third trimester may have more lasting impact on the offspring's propensity to develop obesity than stress encountered at other times. Indeed, this appears to be reflected in the number of elaborate adaptive mechanisms that are present at this time to protect the foetus (Brunton et al. Citation2005, Citation2009; Slattery and Neumann Citation2008; Spencer et al. Citation2008).
Mechanisms for perinatal stress effects on body weight regulation
One mechanism by which prenatal stress (or GC exposure) may result in a long-term susceptibility to obesity is by altering HPA axis function. Prenatal restraint stress leads to altered corticosterone responses to mild stress in the adult rat offspring (Henry et al. Citation1994; Maccari et al. Citation1995, Citation2003; Koehl et al. Citation1999; Rossi-George et al. Citation2009) and reduced hippocampal levels of mineralocorticoid receptor and GR (Henry et al. Citation1994; Maccari et al. Citation1995). Prenatal stress can interfere with synaptic pruning during brain development in regions that are important for HPA axis control, the prefrontal cortex (Spencer et al. Citation2005) and hippocampus (Jacobson and Sapolsky Citation1991). In particular, it has recently been determined that growth-associated protein of 43 kDa (GAP-43), an intracellular protein involved in the establishment and reorganization of synaptic connections during development (Pfenninger et al. Citation1991; Larsson Citation2006), is up-regulated in the second week after birth and reduced in adulthood in offspring from restraint-stressed dams (Jutapakdeegul et al. Citation2010). These findings indicate that prenatal stress can produce lasting changes in the connectivity of these regions, potentially affecting HPA axis sensitivity in the long term. A super-sensitive HPA axis may lead, in turn, to greater susceptibility to the effects of GC on appetite and weight regulation discussed above.
Peripheral alterations in GC function may also come into play. A recent study has determined that 11β-HSD1 may play a crucial role in these programming effects. Thus, chronic prenatal exposure to dexamethasone during late, but not early, gestation led to persistent elevations in 11β-HSD1 mRNA expression and activity in adipose tissue in marmosets. These animals went on to develop early indices of metabolic syndrome (Nyirenda et al. Citation2009).
The mechanisms by which perinatal stress may cause these changes in HPA axis function are not well understood. It is clear that maternal GCs are able to cross the placenta and influence foetal GC levels and receptor expression (Edwards et al. Citation1993) as well as levels of 11β-HSD2 (Clifton et al. Citation2006). Foetal 11β-HSD2 levels are important in programming subsequent stress responses and 11β-HSD2 − / − mice have greater anxiety levels and lower birth weights than wild-type littermates (Holmes et al. Citation2006). Another potential mechanism by which stress may programme later HPA axis function is by DNA methylation. Meaney and colleagues have shown that offspring that were nursed with low intensity (low licking and grooming) develop to be more anxious with exacerbated HPA axis activation in response to stress (Caldji et al. Citation1998; Champagne and Meaney Citation2001). These animals probably have less of a grooming-associated serotonin-mediated increase in nerve growth factor inducible factor A (NGFI-A) expression. NGFI-A binds to its recognition sequence on the GR resulting in increased histone acetylation, which facilitates demethylation of the GR promoter and thus receptor activity. In the case of the low maternal attention groups, this activity would be reduced. The opposite occurs with high maternal attention (Fish et al. Citation2004; Meaney and Szyf Citation2005). Although there is no evidence that these animals develop obesity, such alterations to the GR could predispose the animal to it given high-fat or low-exercise conditions.
Clearly, factors affecting the development of the HPA axis can have long-lasting effects on energy balance within the body. These findings highlight just how important is a stress-free early life environment to long-term body weight control in the offspring.
Perinatal nutritional stress
Inadequate or inappropriate nutrition is a unique type of stressor that has particular long-term impact on the HPA axis and body weight regulation. In the winter of 1944–1945, particularly harsh weather and the final battles and aftermath of Second World War meant that the people of Holland experienced a severe famine. While devastating to the population, several insightful findings on the importance of nutrition have emerged as a result of this disaster. These studies have shown that inadequate nutrition in the first trimester of gestation (but not the last trimester) led to significant obesity in young adult males (Ravelli et al. Citation1976, Citation1999) and middle-aged females. This group also has an altered plasma lipid profile (Roseboom et al. Citation2000a) and a higher risk of coronary heart disease (Roseboom et al. Citation2000b), complications closely associated with obesity. Similar findings have been seen in animal models, with intrauterine growth restriction leading to obesity and related health problems (Jimenez-Chillaron and Patti Citation2007).
As previously suggested, this outcome may be due to a mismatch between the developmental and subsequent environments, with “catch-up growth” leading to an increased susceptibility to obesity. The “predictive-adaptive hypothesis” suggests that an animal adapts specifically to its early life (in utero or immediately post-natal) environment and this may have negative physiological consequences when it is exposed to a different nutritional environment later, i.e. if the “prediction” about the future environment turns out to be inappropriate (Gluckman and Hanson Citation2004; Wadhwa et al. Citation2009). Thus, rapid weight gain in the first weeks or months post partum is a significant risk factor for obesity in later life (Stettler et al. Citation2005). Remarkably, for every 100 g of weight gained in the first week of life, the risk of becoming obese as an adult increases by 28% (Stettler et al. Citation2005). Furthermore, investigation in the rodent has suggested that if this catch-up growth is prevented, the risk of obesity disappears (Ross and Desai Citation2005).
Not surprisingly, perinatal overnutrition is also a risk factor for obesity in later life (McCance Citation1962; Plagemann Citation2006; Chen et al. Citation2008; Morris and Chen Citation2009; Spencer and Tilbrook Citation2009). Although not usually constituting a stressor in itself, perinatal overnutrition does affect the HPA axis (Boullu-Ciocca et al. Citation2005; Spencer and Tilbrook Citation2009), making animals more responsive to stress and potentially contributing to an ongoing cycle of HPA axis hyperactivity and increased weight gain.
Conclusions and future directions
Obesity is currently epidemic within our society and among the many risk factors contributing to the aetiology, stress plays a significant role. Although eliminating stress from our everyday lives is unlikely to occur in the near future, we can work to further our understanding of the mechanisms by which stress may predispose an individual to obesity and potentially target some of these mechanisms.
As discussed, stress affects myriad appetite and adiposity regulating factors that may provide potential targets for treatment of obesity in people with an overactive HPA axis. Ghrelin is one such candidate. Acute and chronic stress both lead to an increase in circulating ghrelin, stimulating food intake (Kristenssson et al. Citation2006; Lutter et al. Citation2008). Ghrelin antagonists or inverse agonists are regarded as promising, but yet unproven, candidates for obesity treatment (Chollet et al. Citation2009). It is possible that these will also ameliorate responses to stress, affecting weight gain by inhibiting GC production as well as via a direct effect on appetite. Another promising target is 11β-HSD1. Mice that do not express 11β-HSD1 are resistant to obesity, and 11β-HSD1 inhibitors have been shown, in rodents, to improve glucose and insulin homeostasis (Morton et al. Citation2004; Stimson and Walker Citation2007; Macfarlane et al. Citation2008; Tiwari Citation2010). Encouragingly, carbenoxolone, a non-specific inhibitor of 11β-HSD1 and 11β-HSD2, has been shown to improve hepatic insulin sensitivity in diabetic and healthy lean patients (Walker et al. Citation1995; Andrews et al. Citation2003). It can also suppress lipolysis in healthy subjects (Tomlinson et al. Citation2007). Clinical trials with specific 11β-HSD1 antagonists are currently underway. Early results report successful inhibition of adipose 11β-HSD1 (Courtney et al. Citation2008), but effects on adiposity and indices of metabolic syndrome remain to be determined (Andrews et al. Citation2003; Fotsch and Wang Citation2008).
Recent critical findings have indicated that the predisposition to obesity in stressed animals may be differentially expressed depending on their sex. For instance, adiposity is differentially distributed in males and females, with males more likely to exhibit visceral adiposity (Hamdy et al. Citation2006). We have shown that female rats, but not males, that were overfed as neonates have enhanced HPA axis responses to psychological stress (Spencer and Tilbrook Citation2009). We also have some recent data to suggest that the mechanism by which obesity is maintained in neonatally overfed rats is different in males and females (Stefanidis, Tilbrook and Spencer unpublished observations, 2009–2010). These findings illustrate that the interplay between responses to stress and subsequent obesity is complex and probably subtly different between the sexes. They also highlight the need for sex-specific treatments for obesity.
The perinatal environment is also crucially important in programming the circuitry that regulates body weight and metabolism. It will be necessary for future studies to define the impact of and identify the mechanisms for stress effects on long-term body weight and metabolism so that we can develop strategies to ameliorate these effects.
Acknowledgements
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia, the Australian Research Council (DP109339), and the Clive and Vera Ramaciotti Foundation. S. J. S. holds a Peter Doherty Fellowship awarded by the NHMRC of Australia (465167) and an Endocrine Society of Australia Postdoctoral Fellowship.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abiles V, Rodriguez-Ruiz S, Abiles J, Mellado C, Garcia A, Perez de la Cruz A, Fernandez-Santaella MC. 2010. Psychological characteristics of morbidly obese candidates for bariatric surgery. Obes Surg. 20:161–167.
- Albiston AL, Smith RE, Krozowski ZS. 1995. Sex- and tissue-specific regulation of 11 beta-hydroxysteroid dehydrogenase mRNA. Mol Cell Endocrinol. 109:183–188.
- Andrews RC, Rooyackers O, Walker BR. 2003. Effects of the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab. 88:285–291.
- Arner P. 2002. Insulin resistance in type 2 diabetes: Role of fatty acids. Diabetes Metab Res Rev. 18 Suppl 2: S5–S9.
- Arvaniti K, Ricquier D, Champigny O, Richard D. 1998. Leptin and corticosterone have opposite effects on food intake and the expression of UCP1 mRNA in brown adipose tissue of lep(ob)/lep(ob) mice. Endocrinology. 139:4000–4003.
- Asarian L, Geary N. 2002. Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav. 42:461–471.
- Asensio C, Muzzin P, Rohner-Jeanrenaud F. 2004. Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int J Obes Relat Metab Disord. 28 Suppl 4: S45–S52.
- Ashby P, Robinson DS. 1980. Effects of insulin, glucocorticoids and adrenaline on the activity of rat adipose-tissue lipoprotein lipids. Biochem J. 188:185–192.
- Baker S, Chebli M, Rees S, Lemarec N, Godbout R, Bielajew C. 2008. Effects of gestational stress: 1. Evaluation of maternal and juvenile offspring behavior. Brain Res. 1213:98–110.
- Balfour ME, Yu L, Coolen LM. 2004. Sexual behavior and sex-associated environmental cues activate the mesolimbic system in male rats. Neuropsychopharmacology. 29:718–730.
- Barry D, Pietrzak RH, Petry NM. 2008. Gender differences in associations between body mass index and DSM-IV mood and anxiety disorders: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Ann Epidemiol. 18:458–466.
- Bassareo V, Di Chiara G. 1997. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci. 17:851–861.
- Bassareo V, Di Chiara G. 1999. Modulation of feeding-induced activation of mesolimbic dopamine transmission by appetitive stimuli and its relation to motivational state. Eur J Neurosci. 11:4389–4397.
- Benson S, Arck PC, Tan S, Mann K, Hahn S, Janssen OE, Schedlowski M, Elsenbruch S. 2009. Effects of obesity on neuroendocrine, cardiovascular, and immune cell responses to acute psychosocial stress in premenopausal women. Psychoneuroendocrinology. 34:181–189.
- Berridge KC. 2009. “Liking” and “wanting” food rewards: Brain substrates and roles in eating disorders. Physiol Behav. 97:537–550.
- Bjorntorp P. 1996. The regulation of adipose tissue distribution in humans. Int J Obes Relat Metab Disord. 20:291–302.
- Bjorntorp P. 2001. Do stress reactions cause abdominal obesity and comorbidities?. Obes Rev. 2:73–86.
- Block JP, He Y, Zaslavsky AM, Ding L, Ayanian JZ. 2009. Psychosocial stress and change in weight among US adults. Am J Epidemiol. 170:181–192.
- Blouin K, Nadeau M, Perreault M, Veilleux A, Drolet R, Marceau P, Mailloux J, Luu-The V, Tchernof A. 2010. Effects of androgens on adipocyte differentiation and adipose tissue explant metabolism in men and women. Clin Endocrinol Oxf. 72:176–188.
- Boullu-Ciocca S, Dutour A, Guillaume V, Achard V, Oliver C, Grino M. 2005. Postnatal diet-induced obesity in rats upregulates systemic and adipose tissue glucocorticoid metabolism during development and in adulthood: Its relationship with the metabolic syndrome. Diabetes. 54:197–203.
- Brunner EJ, Marmot MG, Nanchahal K, Shipley MJ, Stansfeld SA, Juneja M, Alberti KG. 1997. Social inequality in coronary risk: Central obesity and the metabolic syndrome. Evidence from the Whitehall II study. Diabetologia. 40:1341–1349.
- Brunton PJ, Russell JA. 2010. Allopregnanolone and suppressed hypothalamo-pituitary–adrenal axis stress responses in late pregnancy in the rat. Stress, doi. 103109/10253890.2010.482628.
- Brunton PJ, Meddle SL, Ma S, Ochedalski T, Douglas AJ, Russell JA. 2005. Endogenous opioids and attenuated hypothalamic–pituitary–adrenal axis responses to immune challenge in pregnant rats. J Neurosci. 25:5117–5126.
- Brunton PJ, McKay AJ, Ochedalski T, Piastowska A, Rebas E, Lachowicz A, Russell JA. 2009. Central opioid inhibition of neuroendocrine stress responses in pregnancy in the rat is induced by the neurosteroid allopregnanolone. J Neurosci. 29:6449–6460.
- Caldji C, Tannenbaum B, Sharma S, Francis D, Plotsky PM, Meaney MJ. 1998. Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proc Natl Acad Sci USA. 95:5335–5340.
- Cannon B, Nedergaard J. 2004. Brown adipose tissue: Function and physiological significance. Physiol Rev. 84:277–359.
- Champagne F, Meaney MJ. 2001. Like mother, like daughter: Evidence for non-genomic transmission of parental behavior and stress responsivity. Prog Brain Res. 133:287–302.
- Chen H, Simar D, Lambert K, Mercier J, Morris MJ. 2008. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology. 149:5348–5356.
- Chollet C, Meyer K, Beck-Sickinger AG. 2009. Ghrelin-a novel generation of anti-obesity drug: Design, pharmacomodulation and biological activity of ghrelin analogues. J Pept Sci. 15:711–730.
- Ciccocioppo R, Fedeli A, Economidou D, Policani F, Weiss F, Massi M. 2003. The bed nucleus is a neuroanatomical substrate for the anorectic effect of corticotropin-releasing factor and for its reversal by nociceptin/orphanin FQ. J Neurosci. 23:9445–9451.
- Clifton VL, Rennie N, Murphy VE. 2006. Effect of inhaled glucocorticoid treatment on placental 11beta-hydroxysteroid dehydrogenase type 2 activity and neonatal birthweight in pregnancies complicated by asthma. Aust N Z J Obstet Gynaecol. 46:136–140.
- Coccurello R, D'Amato FR, Moles A. 2009. Chronic social stress, hedonism and vulnerability to obesity: Lessons from rodents. Neurosci Biobehav Rev. 33:537–550.
- Commins SP, Watson PM, Padgett MA, Dudley A, Argyropoulos G, Gettys TW. 1999. Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology. 140:292–300.
- Courtney R, Stewart PM, Toh M, Ndongo MN, Calle RA, Hirshberg B. 2008. Modulation of 11beta-hydroxysteroid dehydrogenase. 11betaHSD activity biomarkers and pharmacokinetics of PF-00915275, a selective 11betaHSD1 inhibitor. J Clin Endocrinol Metab. 93:550–556.
- Cummings DE. 2006. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol Behav. 89:71–84.
- Currie PJ. 2003. Integration of hypothalamic feeding and metabolic signals: Focus on neuropeptide Y. Appetite. 41:335–337.
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. 2009. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 360:1509–1517.
- D'Argenio A, Mazzi C, Pecchioli L, Di Lorenzo G, Siracusano A, Troisi A. 2009. Early trauma and adult obesity: Is psychological dysfunction the mediating mechanism?. Physiol Behav. 98:543–546.
- D'Mello AP, Liu Y. 2006. Effects of maternal immobilization stress on birth weight and glucose homeostasis in the offspring. Psychoneuroendocrinology. 31:395–406.
- Dahlgren J, Nilsson C, Jennische E, Ho HP, Eriksson E, Niklasson A, Bjorntorp P, Albertsson WK, Holmang A. 2001. Prenatal cytokine exposure results in obesity and gender-specific programming. Am J Physiol Endocrinol Metab. 281:E326–E334.
- Dallman MF. 2010. Stress-induced obesity and the emotional nervous system. Trends Endocrinol Metab. 21:159–165.
- Dallman MF, la Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF. 2004. Minireview: Glucocorticoids–food intake, abdominal obesity, and wealthy nations in 2004. Endocrinology. 145:2633–2638.
- Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. 2001. Stressor categorization: Acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 14:1143–1152.
- De Pergola G. 2000. The adipose tissue metabolism: Role of testosterone and dehydroepiandrosterone. Int J Obes Relat Metab Disord. 24 Suppl 2: S59–S63.
- De Vriendt T, Moreno LA, De Henauw S. 2009. Chronic stress and obesity in adolescents: Scientific evidence and methodological issues for epidemiological research. Nutr Metab Cardiovasc Dis. 19:511–519.
- Di Chiara G, Imperato A. 1988. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 85:5274–5278.
- Divertie GD, Jensen MD, Miles JM. 1991. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes. 40:1228–1232.
- Doyle AC, le Grange D, Goldschmidt A, Wilfley DE. 2007. Psychosocial and physical impairment in overweight adolescents at high risk for eating disorders. Obesity Silver Spring. 15:145–154.
- Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. 1993. Dysfunction of placental glucocorticoid barrier: Link between fetal environment and adult hypertension?. Lancet. 341:355–357.
- Egecioglu E, Jerlhag E, Salome N, Skibicka KP, Haage D, Bohlooly YM, Andersson D, Bjursell M, Perrissoud D, Engel JA, Dickson SL. 2010. Ghrelin increases intake of rewarding food in rodents. Addict Biol. 15:304–311.
- Entringer S, Buss C, Kumsta R, Hellhammer DH, Wadhwa PD, Wust S. 2009. Prenatal psychosocial stress exposure is associated with subsequent working memory performance in young women. Behav Neurosci. 123:886–893.
- Epel ES, McEwen B, Seeman T, Matthews K, Castellazzo G, Brownell KD, Bell J, Ickovics JR. 2000. Stress and body shape: Stress-induced cortisol secretion is consistently greater among women with central fat. Psychosom Med. 62:623–632.
- Figlewicz DP, Bennett JL, Aliakbari S, Zavosh A, Sipols AJ. 2008. Insulin acts at different CNS sites to decrease acute sucrose intake and sucrose self-administration in rats. Am J Physiol Regul Integr Comp Physiol. 295:R388–R394.
- Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. 2009. Annual medical spending attributable to obesity: Payer-and service-specific estimates. Health Aff Millwood. 28:w822–w831.
- Fish EW, Shahrokh D, Bagot R, Caldji C, Bredy T, Szyf M, Meaney MJ. 2004. Epigenetic programming of stress responses through variations in maternal care. Ann N Y Acad Sci. 1036:167–180.
- Foster MT, Warne JP, Ginsberg AB, Horneman HF, Pecoraro NC, Akana SF, Dallman MF. 2009. Palatable foods, stress, and energy stores sculpt corticotropin-releasing factor, adrenocorticotropin, and corticosterone concentrations after restraint. Endocrinology. 150:2325–2333.
- Fotsch C, Wang M. 2008. Blockade of glucocorticoid excess at the tissue level: Inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 as a therapy for type 2 diabetes. J Med Chem. 51:4851–4857.
- Fowler-Brown AG, Bennett GG, Goodman MS, Wee CC, Corbie-Smith GM, James SA. 2009. Psychosocial stress and 13-year BMI change among blacks: The Pitt County Study. Obesity Silver Spring. 17:2106–2109.
- George SA, Khan S, Briggs H, Abelson JL. 2010. CRH-stimulated cortisol release and food intake in healthy, non-obese adults. Psychoneuroendocrinology. 35:607–612.
- Gluckman PD, Hanson MA. 2004. Developmental origins of disease paradigm: A mechanistic and evolutionary perspective. Pediatr Res. 56:311–317.
- Gosnell BA. 2000. Sucrose intake predicts rate of acquisition of cocaine self-administration. Psychopharmacology Berl. 149:286–292.
- Grassi G, Dell'Oro R, Facchini A, Quarti Trevano F, Bolla GB, Mancia G. 2004. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 22:2363–2369.
- Gregoire F, De Broux N, Hauser N, Heremans H, Van Damme J, Remacle C. 1992. Interferon-gamma and interleukin-1 beta inhibit adipoconversion in cultured rodent preadipocytes. J Cell Physiol. 151:300–309.
- Grove KL, Grayson BE, Glavas MM, Xiao XQ, Smith MS. 2005. Development of metabolic systems. Physiol Behav. 86:646–660.
- Hales CN, Barker DJ. 1992. Type 2. non-insulin-dependent diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia. 35:595–601.
- Hamdy O, Porramatikul S, Al-Ozairi E. 2006. Metabolic obesity: The paradox between visceral and subcutaneous fat. Curr Diabetes Rev. 2:367–373.
- Hauner H, Schmid P, Pfeiffer EF. 1987. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J Clin Endocrinol Metab. 64:832–835.
- Hauner H, Entenmann G, Wabitsch M, Gaillard D, Ailhaud G, Negrel R, Pfeiffer EF. 1989. Promoting effect of glucocorticoids on the differentiation of human adipocyte precursor cells cultured in a chemically defined medium. J Clin Invest. 84:1663–1670.
- Heinrichs SC, Richard D. 1999. The role of corticotropin-releasing factor and urocortin in the modulation of ingestive behavior. Neuropeptides. 33:350–359.
- Heinrichs SC, Menzaghi F, Pich EM, Hauger RL, Koob GF. 1993. Corticotropin-releasing factor in the paraventricular nucleus modulates feeding induced by neuropeptide Y. Brain Res. 611:18–24.
- Henry C, Kabbaj M, Simon H, Le Moal M, Maccari S. 1994. Prenatal stress increases the hypothalamo-pituitary–adrenal axis response in young and adult rats. J Neuroendocrinol. 6:341–345.
- Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. 2006. The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci. 26:3840–3844.
- Hosoda H, Kojima M, Kangawa K. 2006. Biological, physiological, and pharmacological aspects of ghrelin. J Pharmacol Sci. 100:398–410.
- Ito R, Robbins TW, Everitt BJ. 2004. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci. 7:389–397.
- Iwasa T, Matsuzaki T, Kinouchi R, Fujisawa S, Murakami M, Kiyokawa M, Kuwahara A, Yasui T, Irahara M. 2010. Neonatal LPS injection alters the body weight regulation systems of rats under non-stress and immune stress conditions. Int J Dev Neurosci. 28:119–124.
- Jacobson L, Sapolsky R. 1991. The role of the hippocampus in feedback regulation of the hypothalamic–pituitary–adrenocortical axis. Endocr Rev. 12:118–134.
- Jayo JM, Shively CA, Kaplan JR, Manuck SB. 1993. Effects of exercise and stress on body fat distribution in male cynomolgus monkeys. Int J Obes Relat Metab Disord. 17:597–604.
- Jequier E. 2002. Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci. 967:379–388.
- Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, Datta R, Perrissoud D, Dickson SL, Engel JA. 2009. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci USA. 106:11318–11323.
- Jerlhag E, Egecioglu E, Dickson SL, Engel JA. 2010. Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology Berl. 211:415–422.
- Jimenez-Chillaron JC, Patti ME. 2007. To catch up or not to catch up: Is this the question?. Lessons from animal models. Curr Opin Endocrinol Diabetes Obes. 14:23–29.
- Justo R, Frontera M, Pujol E, Rodriguez-Cuenca S, Llado I, Garcia-Palmer FJ, Roca P, Gianotti M. 2005. Gender-related differences in morphology and thermogenic capacity of brown adipose tissue mitochondrial subpopulations. Life Sci. 76:1147–1158.
- Jutapakdeegul N, Polboon N, Afadlal S, Phansuwan-Pujito P, Govitrapong P. 2010. Repeated restraint stress and corticosterone injections during late pregnancy alter GAP-43 expression in the hippocampus and prefrontal cortex of rat pups. Int J Dev Neurosci. 28:83–90.
- Kawakami A, Okada N, Rokkaku K, Honda K, Ishibashi S, Onaka T. 2008. Leptin inhibits and ghrelin augments hypothalamic noradrenaline release after stress. Stress. 11:363–369.
- Keay KA, Bandler R. 2001. Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev. 25:669–678.
- Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. 2005. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes. 54:1023–1031.
- Kinsey-Jones JS, Li XF, Knox AM, Wilkinson ES, Zhu XL, Chaudhary AA, Milligan SR, Lightman SL, O'Byrne KT. 2009. Down-regulation of hypothalamic kisspeptin and its receptor, Kiss1r, mRNA expression is associated with stress-induced suppression of luteinising hormone secretion in the female rat. J Neuroendocrinol. 21:20–29.
- Koch FS, Sepa A, Ludvigsson J. 2008. Psychological stress and obesity. J Pediatr. 153:839–844.
- Koehl M, Darnaudery M, Dulluc J, Van Reeth O, Le Moal M, Maccari S. 1999. Prenatal stress alters circadian activity of hypothalamo-pituitary–adrenal axis and hippocampal corticosteroid receptors in adult rats of both gender. J Neurobiol. 40:302–315.
- Konno J, Yoshida S, Ina A, Ohmomo H, Shutoh F, Nogami H, Hisano S. 2008. Upregulated expression of neuropeptide Y in hypothalamic–pituitary system of rats by chronic dexamethasone administration. Neurosci Res. 60:259–265.
- Koob GF, Le Moal M. 2001. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 24:97–129.
- Kozaric-Kovacic D, Ilic MG, Romic Z, Vidovic A, Jendricko T, Pivac N. 2009. Body mass index in male Caucasian veterans with or without posttraumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry. 33:1447–1450.
- Kristenssson E, Sundqvist M, Astin M, Kjerling M, Mattsson H, Dornonville de la Cour C, Hakanson R, Lindstrom E. 2006. Acute psychological stress raises plasma ghrelin in the rat. Regul Pept. 134:114–117.
- Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, Lee EW, Burnett MS, Fricke ST, Kvetnansky R, Herzog H, Zukowska Z. 2007. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 13:803–811.
- Kuo LE, Czarnecka M, Kitlinska JB, Tilan JU, Kvetnansky R, Zukowska Z. 2008. Chronic stress, combined with a high-fat/high-sugar diet, shifts sympathetic signaling toward neuropeptide Y and leads to obesity and the metabolic syndrome. Ann N Y Acad Sci. 1148:232–237.
- la Fleur SE, Akana SF, Manalo SL, Dallman MF. 2004. Interaction between corticosterone and insulin in obesity: Regulation of lard intake and fat stores. Endocrinology. 145:2174–2185.
- Larsson C. 2006. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 18:276–284.
- Lesage J, Del-Favero F, Leonhardt M, Louvart H, Maccari S, Vieau D, Darnaudery M. 2004. Prenatal stress induces intrauterine growth restriction and programmes glucose intolerance and feeding behaviour disturbances in the aged rat. J Endocrinol. 181:291–296.
- Li J, Olsen J, Vestergaard M, Obel C, Baker JL, Sorensen TI. 2010. Prenatal stress exposure related to maternal bereavement and risk of childhood overweight. PLoS One. 5:e11896.
- Llabre MM, Hadi F. 2009. War-related exposure and psychological distress as predictors of health and sleep: A longitudinal study of Kuwaiti children. Psychosom Med. 71:776–783.
- Lobo MJ, Remesar X, Alemany M. 1993. Effect of chronic intravenous injection of steroid hormones on body weight and composition of female rats. Biochem Mol Biol Int. 29:349–358.
- Lordi B, Protais P, Mellier D, Caston J. 1997. Acute stress in pregnant rats: Effects on growth rate, learning, and memory capabilities of the offspring. Physiol Behav. 62:1087–1092.
- Lovejoy JC, Sainsbury A. 2009. Sex differences in obesity and the regulation of energy homeostasis. Obes Rev. 10:154–167.
- Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. 1993. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 366:740–742.
- Lucassen PJ, Bosch OJ, Jousma E, Kromer SA, Andrew R, Seckl JR, Neumann ID. 2009. Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: Possible key role of placental 11beta-hydroxysteroid dehydrogenase type 2. Eur J Neurosci. 29:97–103.
- Lundgren M, Buren J, Lindgren P, Myrnas T, Ruge T, Eriksson JW. 2008. Sex- and depot-specific lipolysis regulation in human adipocytes: Interplay between adrenergic stimulation and glucocorticoids. Horm Metab Res. 40:854–860.
- Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM. 2008. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 11:752–753.
- Maccari S, Piazza PV, Kabbaj M, Barbazanges A, Simon H, Le Moal M. 1995. Adoption reverses the long-term impairment in glucocorticoid feedback induced by prenatal stress. J Neurosci. 15:110–116.
- Maccari S, Darnaudery M, Morley-Fletcher S, Zuena AR, Cinque C, Van Reeth O. 2003. Prenatal stress and long-term consequences: Implications of glucocorticoid hormones. Neurosci Biobehav Rev. 27:119–127.
- Macfarlane DP, Forbes S, Walker BR. 2008. Glucocorticoids and fatty acid metabolism in humans: Fuelling fat redistribution in the metabolic syndrome. J Endocrinol. 197:189–204.
- Mah PM, Wittert GA. 2010. Obesity and testicular function. Mol Cell Endocrinol. 316:180–186.
- Marin P, Andersson B, Ottosson M, Olbe L, Chowdhury B, Kvist H, Holm G, Sjostrom L, Bjorntorp P. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism. 1992a; 41:1242–1248.
- Marin P, Darin N, Amemiya T, Andersson B, Jern S, Bjorntorp P. Cortisol secretion in relation to body fat distribution in obese premenopausal women. Metabolism. 1992b; 41:882–886.
- Masaki T, Yoshimatsu H, Chiba S, Sakata T. 2000. Impaired response of UCP family to cold exposure in diabetic db/db mice. Am J Physiol Regul Integr Comp Physiol. 279:R1305–R1309.
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. 2001. A transgenic model of visceral obesity and the metabolic syndrome. Science. 294:2166–2170.
- Mathieu P, Pibarot P, Larose E, Poirier P, Marette A, Despres JP. 2008. Visceral obesity and the heart. Int J Biochem Cell Biol. 40:821–836.
- Mayes JS, Watson GH. 2004. Direct effects of sex steroid hormones on adipose tissues and obesity. Obes Rev. 5:197–216.
- McCance RA. 1962. Food, growth, and time. Lancet. 2:671–676.
- Meaney MJ, Szyf M. 2005. Maternal care as a model for experience-dependent chromatin plasticity?. Trends Neurosci. 28:456–463.
- Meijer GA, Janssen GM, Westerterp KR, Verhoeven F, Saris WH, ten Hoor F. 1991. The effect of a 5-month endurance-training programme on physical activity: Evidence for a sex-difference in the metabolic response to exercise. Eur J Appl Physiol Occup Physiol. 62:11–17.
- Moens E, Braet C, Bosmans G, Rosseel Y. 2009. Unfavourable family characteristics and their associations with childhood obesity: A cross-sectional study. Eur Eat Disord Rev. 17:315–323.
- Morley-Fletcher S, Puopolo M, Gentili S, Gerra G, Macchia T, Laviola G. 2004. Prenatal stress affects 3,4-methylenedioxymethamphetamine pharmacokinetics and drug-induced motor alterations in adolescent female rats. Eur J Pharmacol. 489:89–92.
- Morris MJ, Chen H. 2009. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int J Obes Lond. 33:115–122.
- Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR. 2004. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes. 53:931–938.
- Mueller BR, Bale TL. 2006. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 88:605–614.
- Nedergaard J, Bengtsson T, Cannon B. 2007. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 293:E444–E452.
- Nieuwenhuizen AG, Rutters F. 2008. The hypothalamic–pituitary–adrenal-axis in the regulation of energy balance. Physiol Behav. 94:169–177.
- Nilsson C, Larsson BM, Jennische E, Eriksson E, Bjorntorp P, York DA, Holmang A. 2001. Maternal endotoxemia results in obesity and insulin resistance in adult male offspring. Endocrinology. 142:2622–2630.
- Nishitani N, Sakakibara H, Akiyama I. 2009. Eating behavior related to obesity and job stress in male Japanese workers. Nutrition. 25:45–50.
- Nyirenda MJ, Carter R, Tang JI, de Vries A, Schlumbohm C, Hillier SG, Streit F, Oellerich M, Armstrong VW, Fuchs E, Seckl JR. 2009. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11ss-hydroxysteroid dehydrogenase type 1. Diabetes. 58:2873–2879.
- Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. 2006. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 295:1549–1555.
- Papadimitriou A, Priftis KN. 2009. Regulation of the hypothalamic–pituitary–adrenal axis. Neuroimmunomodulation. 16:265–271.
- Pecoraro N, Reyes F, Gomez F, Bhargava A, Dallman MF. 2004. Chronic stress promotes palatable feeding, which reduces signs of stress: Feedforward and feedback effects of chronic stress. Endocrinology. 145:3754–3762.
- Pedersen SB, Kristensen K, Hermann PA, Katzenellenbogen JA, Richelsen B. 2004. Estrogen controls lipolysis by up-regulating alpha2A-adrenergic receptors directly in human adipose tissue through the estrogen receptor alpha. Implications for the female fat distribution. J Clin Endocrinol Metab. 89:1869–1878.
- Perkonigg A, Owashi T, Stein MB, Kirschbaum C, Wittchen HU. 2009. Posttraumatic stress disorder and obesity: Evidence for a risk association. Am J Prev Med. 36:1–8.
- Pfenninger KH, de la Houssaye BA, Helmke SM, Quiroga S. 1991. Growth-regulated proteins and neuronal plasticity. A commentary. Mol Neurobiol. 5:143–151.
- Plagemann A. 2006. Perinatal nutrition and hormone-dependent programming of food intake. Horm Res. 65 Suppl 3: 83–89.
- Ravelli GP, Stein ZA, Susser MW. 1976. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 295:349–353.
- Ravelli AC, Der Meulen JH, Osmond C, Barker DJ, Bleker OP. 1999. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 70:811–816.
- Razzouk L, Muntner P. 2009. Ethnic, gender, and age-related differences in patients with the metabolic syndrome. Curr Hypertens Rep. 11:127–132.
- Rebuffe-Scrive M, Bronnegard M, Nilsson A, Eldh J, Gustafsson JA, Bjorntorp P. 1990. Steroid hormone receptors in human adipose tissues. J Clin Endocrinol Metab. 71:1215–1219.
- Retana-Marquez S, Bonilla-Jaime H, Vazquez-Palacios G, Martinez-Garcia R, Velazquez-Moctezuma J. 2003. Changes in masculine sexual behavior, corticosterone and testosterone in response to acute and chronic stress in male rats. Horm Behav. 44:327–337.
- Richard D, Lin Q, Timofeeva E. 2002. The corticotropin-releasing factor family of peptides and CRF receptors: Their roles in the regulation of energy balance. Eur J Pharmacol. 440:189–197.
- Rodriguez-Cuenca S, Monjo M, Frontera M, Gianotti M, Proenza AM, Roca P. 2007. Sex steroid receptor expression profile in brown adipose tissue. Effects of hormonal status. Cell Physiol Biochem. 20:877–886.
- Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Bleker OP. Plasma lipid profiles in adults after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2000a; 72:1101–1106.
- Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, van Montfrans GA, Michels RP, Bleker OP. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–45. Heart. 2000b; 84:595–598.
- Rosmond R, Dallman MF, Bjorntorp P. 1998. Stress-related cortisol secretion in men: Relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab. 83:1853–1859.
- Ross MG, Desai M. 2005. Gestational programming: Population survival effects of drought and famine during pregnancy. Am J Physiol Regul Integr Comp Physiol. 288:R25–R33.
- Rossi-George A, Virgolini MB, Weston D, Cory-Slechta DA. 2009. Alterations in glucocorticoid negative feedback following maternal Pb, prenatal stress and the combination: A potential biological unifying mechanism for their corresponding disease profiles. Toxicol Appl Pharmacol. 234:117–127.
- Ryu V, Yoo SB, Kang DW, Lee JH, Jahng JW. 2009. Post-weaning isolation promotes food intake and body weight gain in rats that experienced neonatal maternal separation. Brain Res. 1295:127–134.
- Saad F, Gooren L. 2009. The role of testosterone in the metabolic syndrome: A review. J Steroid Biochem Mol Biol. 114:40–43.
- Santana P, Akana SF, Hanson ES, Strack AM, Sebastian RJ, Dallman MF. 1995. Aldosterone and dexamethasone both stimulate energy acquisition whereas only the glucocorticoid alters energy storage. Endocrinology. 136:2214–2222.
- Sapolsky RM, Romero LM, Munck AU. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 21:55–89.
- Sartorio A, Maffiuletti NA, Agosti F, Lafortuna CL. 2005. Gender-related changes in body composition, muscle strength and power output after a short-term multidisciplinary weight loss intervention in morbid obesity. J Endocrinol Invest. 28:494–501.
- Savontaus E, Conwell IM, Wardlaw SL. 2002. Effects of adrenalectomy on AGRP, POMC, NPY and CART gene expression in the basal hypothalamus of fed and fasted rats. Brain Res. 958:130–138.
- Schmidt MV, Czisch M, Sterlemann V, Reinel C, Samann P, Muller MB. 2009. Chronic social stress during adolescence in mice alters fat distribution in late life: Prevention by antidepressant treatment. Stress. 12:89–94.
- Scott KM, McGee MA, Wells JE, Oakley Browne MA. 2008. Obesity and mental disorders in the adult general population. J Psychosom Res. 64:97–105.
- Shi H, Seeley RJ, Clegg DJ. 2009. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol. 30:396–404.
- Shimizu H, Arima H, Watanabe M, Goto M, Banno R, Sato I, Ozaki N, Nagasaki H, Oiso Y. 2008. Glucocorticoids increase neuropeptide Y and agouti-related peptide gene expression via adenosine monophosphate-activated protein kinase signaling in the arcuate nucleus of rats. Endocrinology. 149:4544–4553.
- Shively CA. 1998. Social subordination stress, behavior, and central monoaminergic function in female cynomolgus monkeys. Biol Psychiatry. 44:882–891.
- Shively CA, Clarkson TB. 1988. Regional obesity and coronary artery atherosclerosis in females: A non-human primate model. Acta Med Scand Suppl. 723:71–78.
- Shively CA, Register TC, Clarkson TB. 2009. Social stress, visceral obesity, and coronary artery atherosclerosis in female primates. Obesity Silver Spring. 17:1513–1520.
- Slattery DA, Neumann ID. 2008. No stress please! Mechanisms of stress hyporesponsiveness of the maternal brain. J Physiol. 586:377–385.
- Slavin BG, Ong JM, Kern PA. 1994. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res. 35:1535–1541.
- Spencer SJ, Tilbrook A. 2009. Neonatal overfeeding alters adult anxiety and stress responsiveness. Psychoneuroendocrinology. 34:1133–1143.
- Spencer SJ, Buller KM, Day TA. 2005. Medial prefrontal cortex control of the paraventricular hypothalamic nucleus response to psychological stress: Possible role of the bed nucleus of the stria terminalis. J Comp Neurol. 481:363–376.
- Spencer SJ, Mouihate A, Galic MA, Ellis SL, Pittman QJ. 2007. Neonatal immune challenge does not affect body weight regulation in rats. Am J Physiol Regul Integr Comp Physiol. 293:R581–R589.
- Spencer SJ, Mouihate A, Galic MA, Pittman QJ. 2008. Central and peripheral neuroimmune responses: Hyporesponsiveness during pregnancy. J Physiol. 586:399–406.
- Stettler N, Stallings VA, Troxel AB, Zhao J, Schinnar R, Nelson SE, Ziegler EE, Strom BL. 2005. Weight gain in the first week of life and overweight in adulthood: A cohort study of European American subjects fed infant formula. Circulation. 111:1897–1903.
- Stimson RH, Walker BR. 2007. Glucocorticoids and 11beta-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Minerva Endocrinol. 32:141–159.
- Strack AM, Sebastian RJ, Schwartz MW, Dallman MF. 1995. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am J Physiol Regul Integr Comp Physiol. 268:R142–R149.
- Suryawan A, Swanson LV, Hu CY. 1997. Insulin and hydrocortisone, but not triiodothyronine, are required for the differentiation of pig preadipocytes in primary culture. J Anim Sci. 75:105–111.
- Svendsen OL, Hassager C, Christiansen C. 1995. Age- and menopause-associated variations in body composition and fat distribution in healthy women as measured by dual-energy X-ray absorptiometry. Metabolism. 44:369–373.
- Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH. 2009. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes. 58:1116–1125.
- Tchernof A, Poehlman ET, Despres JP. 2000. Body fat distribution, the menopause transition, and hormone replacement therapy. Diabetes Metab. 26:12–20.
- Thomas MB, Hu M, Lee TM, Bhatnagar S, Becker JB. 2009. Sex-specific susceptibility to cocaine in rats with a history of prenatal stress. Physiol Behav. 97:270–277.
- Tiwari A. 2010. INCB-13739, an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor for the treatment of type 2 diabetes. IDrugs. 13:266–275.
- Tomlinson JW, Stewart PM. 2002. The functional consequences of 11beta-hydroxysteroid dehydrogenase expression in adipose tissue. Horm Metab Res. 34:746–751.
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 2004. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr Rev. 25:831–866.
- Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, Courtney R, Rejto P, Carley W, Stewart PM. 2007. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab. 92:857–864.
- Toogood AA, Taylor NF, Shalet SM, Monson JP. 2000. Sexual dimorphism of cortisol metabolism is maintained in elderly subjects and is not oestrogen dependent. Clin Endocrinol Oxf. 52:61–66.
- Valle A, Catala-Niell A, Colom B, Garcia-Palmer FJ, Oliver J, Roca P. 2005. Sex-related differences in energy balance in response to caloric restriction. Am J Physiol Endocrinol Metab. 289:E15–E22.
- Valle A, Garcia-Palmer FJ, Oliver J, Roca P. 2007. Sex differences in brown adipose tissue thermogenic features during caloric restriction. Cell Physiol Biochem. 19:195–204.
- Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. 1997. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: Correlation with stress-induced corticosterone secretion. J Neurosci. 17:2626–2636.
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. 2009. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 360:1500–1508.
- Veilleux A, Rheaume C, Daris M, Luu-The V, Tchernof A. 2009. Omental adipose tissue type 1 11 beta-hydroxysteroid dehydrogenase oxoreductase activity, body fat distribution, and metabolic alterations in women. J Clin Endocrinol Metab. 94:3550–3557.
- Vicennati V, Pasqui F, Cavazza C, Pagotto U, Pasquali R. 2009. Stress-related development of obesity and cortisol in women. Obesity Silver Spring. 17:1678–1683.
- Vieweg WV, Julius DA, Bates J, Quinn JF3rd, Fernandez A, Hasnain M, Pandurangi AK. 2007. Posttraumatic stress disorder as a risk factor for obesity among male military veterans. Acta Psychiatr Scand. 116:483–487.
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, Nuutila P. 2009. Functional brown adipose tissue in healthy adults. N Engl J Med. 360:1518–1525.
- Wadhwa PD, Buss C, Entringer S, Swanson JM. 2009. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 27:358–368.
- Waite M, Kershaw Y, Lightman SL. 2009. A glucocorticoid sensitive biphasic rhythm of testosterone secretion. J. Neuroendo. 21:737–741.
- Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. 1995. Carbenoxolone increases hepatic insulin sensitivity in man: A novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. J Clin Endocrinol Metab. 80:3155–3159.
- Warne JP, Horneman HF, Wick EC, Bhargava A, Pecoraro NC, Ginsberg AB, Akana SF, Dallman MF. 2006. Comparison of superior mesenteric versus jugular venous infusions of insulin in streptozotocin-diabetic rats on the choice of caloric intake, body weight, and fat stores. Endocrinology. 147:5443–5451.
- Warne JP, Akana SF, Ginsberg AB, Horneman HF, Pecoraro NC, Dallman MF. 2009. Disengaging insulin from corticosterone: Roles of each on energy intake and disposition. Am J Physiol Regul Integr Comp Physiol. 296:R1366–R1375.
- Weaver JU, Taylor NF, Monson JP, Wood PJ, Kelly WF. 1998. Sexual dimorphism in 11 beta hydroxysteroid dehydrogenase activity and its relation to fat distribution and insulin sensitivity; a study in hypopituitary subjects. Clin Endocrinol Oxf. 49:13–20.
- Weninger SC, Muglia LJ, Jacobson L, Majzoub JA. 1999. CRH-deficient mice have a normal anorectic response to chronic stress. Regul Pept. 84:69–74.
- Westerterp KR, Goran MI. 1997. Relationship between physical activity related energy expenditure and body composition: A gender difference. Int J Obes Relat Metab Disord. 21:184–188.
- Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, Xu G. 2009. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol Endocrinol. 23:1161–1170.
- Zakrzewska KE, Cusin I, Sainsbury A, Rohner-Jeanrenaud F, Jeanrenaud B. 1997. Glucocorticoids as counterregulatory hormones of leptin: Toward an understanding of leptin resistance. Diabetes. 46:717–719.
- Zakrzewska KE, Cusin I, Stricker-Krongrad A, Boss O, Ricquier D, Jeanrenaud B, Rohner-Jeanrenaud F. 1999. Induction of obesity and hyperleptinemia by central glucocorticoid infusion in the rat. Diabetes. 48:365–370.
- Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, Nedergaard J, Cinti S. 2009. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. Faseb J. 23:3113–3120.