Abstract
Endogenous cannabinoids play an important role in the physiology and behavioral expression of stress responses. Activation of the hypothalamic–pituitary–adrenal (HPA) axis, including the release of glucocorticoids, is the fundamental hormonal response to stress. Endocannabinoid (eCB) signaling serves to maintain HPA-axis homeostasis, by buffering basal activity as well as by mediating glucocorticoid fast feedback mechanisms. Following chronic stressor exposure, eCBs are also involved in physiological and behavioral habituation processes. Behavioral consequences of stress include fear and stress-induced anxiety as well as memory formation in the context of stress, involving contextual fear conditioning and inhibitory avoidance learning. Chronic stress can also lead to depression-like symptoms. Prominent in these behavioral stress responses is the interaction between eCBs and the HPA-axis. Future directions may differentiate among eCB signaling within various brain structures/neuronal subpopulations as well as between the distinct roles of the endogenous cannabinoid ligands. Investigation into the role of the eCB system in allostatic states and recovery processes may give insight into possible therapeutic manipulations of the system in treating chronic stress-related conditions in humans.
Introduction
The endocannabinoid system
Cannabis sativa and its derivatives have been used therapeutically and recreationally throughout the world for millennia; however, until relatively recently, the mechanisms mediating their effects remained largely unknown. It was not until 1964 that Δ-9-tetrahydrocannabinol (Δ9-THC) was isolated and identified as the main psychoactive component of cannabis (Goani and Mechoulam Citation1964). It was initially assumed that the effects of Δ9-THC were mediated through nonspecific mechanisms, such as modulation of membrane fluidity; but, in the late 1980s, it was elucidated that cannabinoids exert their effects by acting upon a membrane-bound, G-protein coupled receptor (Hillard et al. Citation1985; Devane et al. Citation1988). Shortly thereafter, the cannabinoid type 1 (CB1) receptor was genetically identified and cloned (Matsuda et al. Citation1990). It was found to be widely distributed throughout most regions in the brain and is considered the most abundant G-protein coupled receptor in the central nervous system (Devane et al. Citation1988; Herkenham et al. Citation1990, Citation1991; Matsuda et al. Citation1990).
CB1 receptors are localized presynaptically and their activation functions to inhibit adenylate cyclase as well as to activate potassium channels and inhibit N- and P/Q-type calcium channels; these are actions which all serve to reduce excitability of the presynaptic neuron and inhibit neurotransmitter release (Howlett and Fleming Citation1984; Howlett Citation1987; Mackie and Hille Citation1992; Mackie et al. Citation1995; Twitchell et al. Citation1997). A second cannabinoid receptor, the CB2 receptor, has also been identified (Munro et al. Citation1993). The CB2 receptor is a G-protein coupled receptor and shares some signaling characteristics with the CB1 receptor, although its distinct and limited localization within the brain indicates that it regulates processes that are different from those regulated by the CB1 receptor (Van Sickle et al. Citation2005).
Endogenous ligands that bind cannabinoid receptors, collectively referred to as endocannabinoids (eCBs), were identified not long after the discovery of the CB1 receptor. The most predominant and widely investigated ligands are the arachidonate-derived molecules, N-arachidonylethanolamine (anandamide; AEA) and 2-arachidonoylglycerol (2-AG; Devane et al. Citation1992; Sugiura et al. Citation1995). However, several other endogenous molecules have been found to bind cannabinoid receptors, such as noladin ether, oleamide, and virodhamine (Hanus et al. Citation2001; Porter et al. Citation2002; Leggett et al. Citation2004). This review will focus on the endogenous signaling molecules AEA and 2-AG, as their role in stress has been more thoroughly investigated than that of other CB1 ligands.
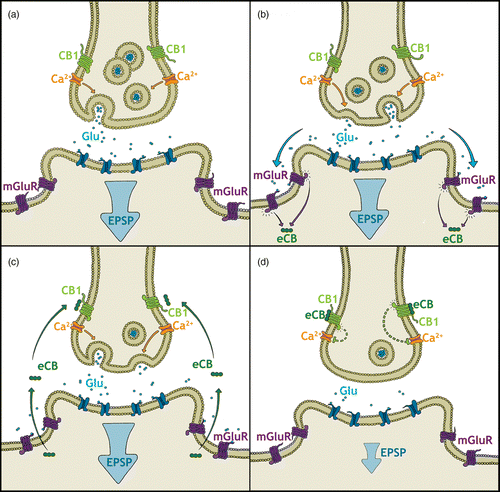
Both AEA and 2-AG are produced postsynaptically in a Ca2+-dependent manner following neuronal depolarization as well as upon activation of metabotropic receptors and stimulation of secondary messenger mechanisms (Di Marzo et al. Citation1994; Stella et al. Citation1997; Piomelli Citation2003). Rather than being stored in vesicles like classical neurotransmitters, eCBs are produced “on demand” following depolarization of the postsynaptic neuron (Piomelli Citation2003). The biosynthesis of 2-AG is mediated by the enzymes phospholipase C and diacylglycerol lipases, but several possible pathways seem to contribute to the synthesis of AEA (Stella et al. Citation1997; Bisogno Citation2008). Upon synthesis, eCBs cross the synapse in a retrograde fashion toward the presynaptically located cannabinoid receptors. provides an exemplary model of retrograde eCB signaling. Several lines of evidence indicate the existence of an eCB transporter to assist these hydrophobic molecules in crossing cellular membranes, and although specific transport inhibitors have been produced, no transport protein has been identified (Lovinger Citation2007). eCB signaling is terminated through reuptake mechanisms and enzymatic degradation; AEA is hydrolyzed by postsynaptic, membrane-bound fatty acid amide hydrolase (FAAH), whereas 2-AG is predominantly hydrolyzed by presynaptically localized monoacylglycerol lipase (Cravatt et al. Citation1996; Egertová et al. Citation1998; Goparaju et al. Citation1999; Dinh et al. Citation2002). Altogether, the eCB system encompasses the cannabinoid receptor and its endogenous ligands as well as the enzymatic machinery involved in the synthesis and degradation of these ligands and any putative transport molecules that may assist in their movement within and between cells.
Figure 1. A hypothetical model of retrograde eCB signaling at a glutamatergic synapse. (a) Glutamate (Glu) is released at moderate levels activating postsynaptic ionotropic glutamate receptors leading to an excitatory postsynaptic potential (EPSP). (b) Increased or prolonged glutamate release additionally activates G-protein coupled metabotropic glutamate receptors (mGluR). A secondary messenger cascade leads to the synthesis of eCB. (c) Newly synthesized eCBs travel back across the synapse to activate G-protein coupled CB1 receptors. (d) CB1 receptor activation inhibits further neurotransmitter release by blocking calcium Ca2+ influx into the presynaptic cell among other actions. Glutamate release is downregulated and the EPSP is attenuated (figure modified from Moreira and Wotjak Citation2010).
Stress and stressors
Stress for biological organisms is most fundamentally described as a state of strain due to threatened homeostasis (Cannon Citation1935). Selye (Citation1950) first made the important distinction between stressors and stress, with stressors defined as stimuli and stress as the internal state experienced by the organism upon exposure to a stressor. Upon coining the term “homeostasis,” Cannon (Citation1935) referred to the relatively nonspecific physiological consequences of exposure to physical stressors such as cold, hypoxia, hypoglycemia, and hemorrhage. In contrast, some potential stressors require the individual to evaluate, through processing by neural circuitry, their own coping abilities and resources in the face of an environmental demand to determine whether a stimulus is indeed a stressor (Lazarus Citation1966; Lazarus and Launier Citation1978; Lazarus and Folkman Citation1984). Psychological stressors may be perceived as stressful to the individual even though they do not directly disrupt physiological homeostasis. Herman et al. (Citation2003) referred to physical stressors as “reactive” stressors, since they cause a direct disruption in homeostatic functioning, and psychological stressors as “anticipatory” stressors, since they only anticipate a homeostatic disruption based on either prior experiences or species-specific predispositions. Neural input for physical stressors originate from the spinal cord and, in particular, from hindbrain regions, which receive input from sensory fibers in the periphery, whereas psychological stressors depend on forebrain inputs from areas such as the prefrontal cortex (PFC) and hippocampus (HPC), which are necessary to evaluate the nature of a potential stressor (Herman et al. Citation2003). Stressors such as forced swimming, common in preclinical research, have a psychological component to them, but also relatively strong and direct physical consequences as well (i.e. lowered body temperature and increased energy consumption, respectively). Stressors such as restraint, predator odor, or social defeat contain some physical elements to them, but are more psychological or “anticipatory” in nature. The current review focuses on stressors with stronger psychological vs. physical components in relation to eCB system functioning.
Experimental stress protocols conventionally differentiate between chronic and acute paradigms; however, when duration and frequency of the stressor exposure are taken into account, one can further distinguish among several types of chronic stressors as well. An acute protocol is a relatively brief one-time exposure to a single stressor; whereas, a chronic protocol can be either a prolonged one-time exposure to a single stressor or a repeated exposure to either the same (homotypic) or variable (heterotypic) stressor(s). An example of a commonly used heterotypic, repeated stressor is the chronic mild stress protocol (CMS; Willner et al. Citation1987; Willner Citation1997). Chronic stressor exposure typically involves some form of habituation, which serves to dampen the detrimental effects of a prolonged neurochemical stress response as well as increase the psychological tolerability of the stressor. Discriminating among different types of chronic stressors is important for understanding the habituation processes that can occur. For example, habituation of physiological and behavioral processes to repeated heterotypic stressors, such as those presented in the CMS protocol, is typically quite limited or nonexistent (Willner Citation1997). In addition, habituation processes occurring in response to repeated homotypic stressors can be stressor specific as well as dependent upon the length of the interstressor interval and intensity of the stressor (Kant et al. Citation1985; Natelson et al. Citation1988; De Boer et al. Citation1990). Furthermore, habituation occurring within a single stressor exposure may be distinct from the habituation that occurs between subsequent stressor exposures.
Endocannabinoids and the hormonal stress response
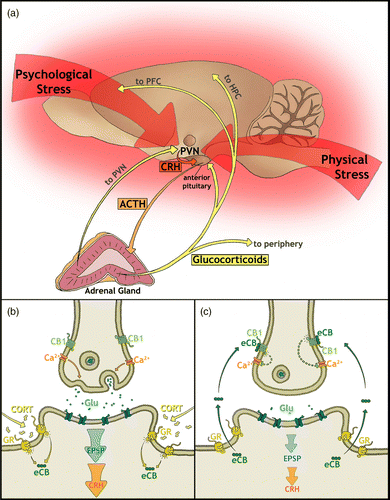
Investigation of eCB regulation of hormonal stress responses has only recently begun; however, it is already clear that they play a pivotal role in restoring hormonal homeostatic functioning following stress. One of the most fundamental physiological responses to stress is activation of the hypothalamic–pituitary–adrenal (HPA) axis (). Stress-induced neuronal stimulation results in activation of parvocellular neurosecretory cells in the paraventricular nucleus (PVN) of the hypothalamus, which synthesize and release corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP; de Kloet et al. Citation2005; Pecoraro et al. Citation2006). Via the median eminence and hypophyseal portal system, CRH and AVP are transported to the anterior pituitary leading to the release of adrenocorticotropic hormone (ACTH) into the bloodstream. Upon reaching the adrenal gland, ACTH stimulates the release of glucocorticoids from the adrenal cortex, which act on peripheral and neuronal glucocorticoid receptors (GR; de Kloet et al. Citation2005; Pecoraro et al. Citation2006). Hormonal stress responses, such as activation of the HPA-axis, serve to prepare an organism for possible threat by mobilizing energy, increasing arousal, focusing attention, and mediating cognition while dampening digestive, immune, and reproductive functions (Chrousos and Gold Citation1992; Tsigos and Chrousos Citation2002).
Figure 2. The HPA-axis and its glucocorticoid feedback mechanisms including exemplary eCB-mediated negative feedback in the PVN. (a) Neuronal input into the HPA-axis from psychological stressors originates from cortical-based cognitive and decision-making brain areas, whereas input from physical stressors is from lower hindbrain regions (Herman et al. Citation2003). These inputs culminate in the activation of neurosecretory cells in the PVN and the release of CRH. In the anterior pituitary, CRH stimulates the release of ACTH into circulation where it activates glucocorticoid production in the adrenal cortex. Glucocorticoids travel to the periphery to have widespread physiological effects as well as providing negative feedback to the HPA-axis via several brain areas, including the PVN, PFC, and hippocampus (HPC; de Kloet et al. Citation2005; Pecoraro et al. Citation2006). (b) Glucocorticoid fast feedback in the PVN stimulates membrane-associated GRs, which in turn leads to the synthesis of (eCBs; Di et al. Citation2003; Tasker et al. Citation2006). (c) eCBs activate presynaptically located CB1 receptors to inhibit further neurotransmitter release. Glutamatergic activation of EPSPs in the postsynaptic neurosecretory cell is reduced, subsequently leading to decreased CRH release and downregulation of HPA-axis activity (Di et al. Citation2003; Tasker et al. Citation2006).
Tonic eCB signaling evidently plays a role in constraining HPA-axis activity, and downregulation of eCB signaling may be important for induction of the hormonal stress response. It is theorized that tonic AEA levels serve as a “gatekeeper” of HPA-axis functioning, which must be lowered to allow for stress-induced activation of the HPA-axis response (Hill et al. Citation2009b). Enhancement of eCB signaling dampens stress-induced HPA-axis activity, and blockade of CB1 receptors has been shown to cause an increase in stress-induced ACTH and corticosterone release as well as an increased neuronal activation in the PVN, all indicative of increased HPA-axis functioning (Patel et al. Citation2004; Steiner and Wotjak Citation2008; Ginsberg et al. Citation2010). However, CB1 antagonism directly in the PVN does not affect stress-induced ACTH release (Evanson et al. Citation2010), indicating that the above-mentioned effects are not due to CB1 blockade directly in the PVN. Alternatively, systemic CB1 blockade could preferentially result in disinhibition of excitatory inputs into the PVN causing activation of the PVN and subsequently of the HPA-axis. A likely candidate for such an input region is the amygdala, which activates the HPA-axis and exhibits stress-induced decreases in AEA content that correlate negatively with corticosterone secretion (Herman et al. Citation2005; Patel et al. Citation2005; Rademacher et al. Citation2008; Hill et al. Citation2009b). Although the effect of restraint stress is dependent on downregulation of AEA in the basolateral amygdalar, the role of amygdalar eCB signaling in conditioned fear may rely on distinct neuronal networks (Hill et al. Citation2009b; Kamprath et al. Citation2010).
To avoid unnecessary catabolic and immunosuppressive effects of protracted glucocortocoid secretion, negative feedback mechanisms are in place to regulate HPA-axis activity. eCB signaling has been shown to play an integral part in this feedback process. Nongenomic or fast feedback of the HPA-axis occurs via glucocorticoid suppression of ACTH release in the pituitary and of CRH release in the hypothalamus (Keller-Wood and Dallman Citation1984; Hinz and Hirschelmann Citation2000; Pecoraro et al. Citation2006). Although classical GR signaling depends on activation of intracellular receptors mediating gene transcription, fast feedback mechanisms are assumed to occur via nongenomic mechanisms that rely on membrane-associated GR receptors (Tasker et al. Citation2006; Tasker and Herman Citation2011). This rapid GR feedback on CRH-releasing neurons within the PVN has been shown to be mediated by retrograde eCB inhibition of glutamate release in vitro (Di et al. Citation2003). eCB signaling has also been shown to be necessary for stress-induced inhibition of ACTH and corticosterone release in vivo (Evanson et al. Citation2010). Increases in hypothalamic 2-AG and AEA content following restraint stress or glucocorticoid treatment support the notion that eCB synthesis is induced in the PVN by glucocorticoid feedback, which inhibits further activation of CRH cells and dampens HPA-axis activity (; Malcher-Lopes et al. Citation2006; Evanson et al. Citation2010; Hill et al. Citation2010a). In this way, eCBs help mediate the reinstatement of homeostatic functioning following stress-induced disruption of HPA-axis homeostasis.
Exposure to repeated stressors can lead to long-lasting physiological and behavioral changes, both of which can be adaptive or maladaptive. Evidence indicates that the eCB system plays an integral role in the development of physiological habituation, and amygdalar eCB signaling is emerging as a key neuronal mechanism in these processes. Repeated restraint has been consistently shown to simultaneously decrease AEA and increase 2-AG content in the amygdalar (Patel et al. Citation2005; Rademacher et al. Citation2008; Hill et al. Citation2010b). Amygdalar decreases in AEA signaling have been speculated to be responsible for increased basal secretion of glucocorticoid following repeated stressor exposure, whereas the enhanced 2-AG activity may contribute to HPA-axis habituation upon subsequent stressor exposure (Hill et al. Citation2010b). However, elevated levels of 2-AG could indicate a loss of inhibition in the amygdala (Patel et al. Citation2009); therefore, behavioral measurements are needed to confirm the effects of these changes on emotionality. Similarly, downregulation of eCB signaling in the PVN following repeated stress (Hill et al. Citation2010b; Wamsteeker et al. Citation2010) could indicate a loss of inhibitory feedback of the HPA-axis; however, these changes have yet to be assessed behaviorally. For example, following CMS, in which habituation typically does not occur (Willner Citation1997), loss of eCB signaling in the HPC has been related to cognitive impairment (Hill et al. Citation2005). Changes in eCB signaling following repeated stress may help habituate HPA-axis activity and protect against the deleterious effects of long-term HPA-axis activation and chronically elevated glucocorticoid, which may include cognitive impairments, psychological disturbances, and development of severe chronic diseases (Seyle Citation1950; Chrousos and Gold Citation1992; Bodnoff et al. Citation1995; Checkley Citation1996; de Kloet et al. Citation2005). However, dysregulation of eCB signaling following chronic stress may be related to such detrimental effects and it remains to be determined whether the observed changes in eCB system functioning are adaptive or not. Future research may also need to distinguish between changes in eCB signaling which occur following repeated stress in animals resistance vs. susceptible to the detrimental behavioral effects of chronic stress.
Endocannabinoids and behavioral stress responses
The interaction between a stressor exposure and the eCB system may affect psychoemotional and cognitive behavior of animals in terms of conditioned fear, anxiety, and memory processes. In addition, chronic stressor exposure may lead to depression-like symptoms, which are also modulated by eCBs. Other more reflexive behaviors affected by stress and regulated by eCBs include pain perception, in the case of stress-induced analgesia, and sexual behavior. The phenomenon of analgesia and its relation to eCB system functioning has been more extensively reviewed in a number of previous publications (Hohmann and Suplita Citation2006; Ford and Finn Citation2008; Butler and Finn Citation2009; Finn Citation2010) and, therefore, will not be covered here. However, eCB-mediation of stress-induced sexual behaviors has only been investigated in the rough-skinned newt (Coddington et al. Citation2007) and has yet to be extended to mammals. Instead, this article focuses on psychoemotional and cognitive behaviors in the context of animal models, and in relation to stress-related psychiatric conditions in humans. Although other excellent reviews on the interaction between the eCB system and anxiety, fear, memory processes and depression can be found elsewhere (Finn Citation2010), we focus here on these interactions specifically in the context of stress exposure. It cannot be in the scope of the present article to discuss all of these interactions in great detail. Instead, the following paragraphs provide some examples illustrating the complexity of eCB action in these aspects.
Stress and fear
Pharmacological blockade of CB1 receptors causes sustained behavioral arrest (i.e. freezing) and prolonged ultrasound vocalization upon re-exposure to the aversive conditioning context (Finn et al. Citation2004). The role of the eCB system in fear adaptation has been analyzed in detail in response to aversive acoustic stimuli. For instance, presentation of a sine wave tone may cause freezing: if a loud tone is presented to naïve animals, or if a tone of intermediate intensity is presented to mice that had received an unsignaled electric foot shock 24 h before, or if the tone was associated with the foot shock during Pavlovian conditioning (Kamprath and Wotjak Citation2004). In all cases, the freezing response decreases over the course of tone presentation, likely because of habituation-like processes (Kamprath and Wotjak Citation2004). Accordingly, pharmacological or genetic interruption of CB1 signaling leads to sustained fear responses over the course of the tone presentation (Kamprath et al. Citation2006; Niyuhire et al. Citation2007; Plendl and Wotjak Citation2010). However, the role of the eCB system in short-term habituation becomes apparent only in highly aversive situations (Kamprath et al. Citation2009). Such acute fear relief involves CB1-mediated control of amygdalar glutamatergic neurons, likely independently of potential changes in HPA-axis activity, within both the basolateral and central amygdala (Kamprath et al. Citation2009, Citation2010).
A recent study investigated the interaction between stressor exposure, glucocorticoids, CB1 signaling within the dorsal HPC, and acquisition/consolidation of contextual fear memory (de Oliveira Alvares et al. Citation2010). Intra-hippocampal administration of a CB1 receptor antagonist inhibited the formation of contextual fear if administered after conditioning with a strong, but not with a weak foot shock. The eCB-independent fear memory formation after conditioning with the weak foot shock became eCB dependent, if conditioning was preceeded by a stressor exposure (electric foot shocks in a different context) or systemic treatment with a synthetic glucocorticoid. This effect could be observed only if stressor exposure/treatment occurred directly before conditioning, but not 30 min earlier. Even though more experimental evidence is necessary, these data indicate that increased secretion of glucocorticoids leads to a transient activation of eCB signaling within the dorsal HPC, which in turn exerts facilitating effects on fear memory.
It has been found that CB1 deficiency or antagonism leads to decreased contextual fear, but to impaired extinction of conditioned fear responses (Marsicano et al. Citation2002; Chhatwal et al. Citation2005; Mikics et al. Citation2006; de Oliveira Alvares et al. Citation2010). However, it has also been found that pharmacological enhancement of eCB signaling enhances the extinction of conditioned as well as contextual fear (Chhatwal et al. Citation2005; Bitencourt et al. Citation2008; Pamplona et al. Citation2008), which would suggest a differential role of eCB signaling in the acquisition vs. expression of contextual fear. Posttraumatic stress disorder and phobias are conditions in which stressor exposure can cause long-lasting fear in relation to the stressor context and to stimuli conditioned to the stressor, among other symptoms. The long-term reduction in contextual fear via enhancement of CB1 signaling (Pamplona et al. Citation2008) could indicate a therapeutic possibility in the treatment of posttraumatic stress disorder or phobias.
Stress and memory processes
A stressful encounter may affect memory processes outside those involved in conditioned fear memory. For instance, acute exposure of rats to an elevated platform increases acquisition/consolidation and attenuates extinction in an inhibitory avoidance task (Ganon-Elazar and Akirav Citation2009). These stress effects could be blocked by injection of a CB1 receptor agonist into the basolateral amygdala, which also attenuated stress-induced corticosterone secretion (Ganon-Elazar and Akirav Citation2009). Conversely, systemic injection of corticosterone improved inhibitory avoidance memory, which could be blocked by injection of a CB1 receptor antagonist into the basolateral amygdala (Campolongo et al. Citation2009). Future studies have to resolve the conundrum that both activation and inhibition of CB1 signaling seem to inhibit the formation of inhibitory avoidance memories. In this context, attention should be paid to dissociations in the effects of stressor exposure vs. corticosterone injection on activation of eCB signaling (Hill et al. Citation2010a,Citationb). Moreover, future investigations might focus on the interaction between eCB signaling and noradrenergic-mediated memory consolidation, which has been proposed as a possible mechanism within the basolateral amygdala (Hill and McEwen Citation2009).
Other studies have evaluated memory processes following chronic stress, such as CMS exposure. Chronic mild stressor exposure was found to reduce CB1 expression and binding as well as 2-AG content in the HPC, which was paralleled by cognitive deficits in the Morris water maze (MWM) task (Hill et al. Citation2005). These deficits were specific to reversal trials, in which the platform location was moved, and did not affect initial acquisition learning. Although these results may suggest a positive relationship between loss of CB1 signaling and cognitive decline, fear learning in an active avoidance task following CMS exposure was enhanced in CB1-deficient mice (Martin et al. Citation2002). The cognitive impairments seen in the MWM task (Hill et al. Citation2005) may relate to the finding that CB1 deficiency or antagonism results in normal learning, but impaired memory extinction, similar to the reduction in extinction of fear memories (Marsicano et al. Citation2002). The impaired memory extinction with CB1 downregulation may relate not only to the inability of rats to relearn during the MWM task, but also to the increased retention (i.e. decreased extinction) of fear memory seen in the inhibitory avoidance task. Interestingly, eCB signaling is evidently specific to the extinction of aversive, but not appetitive memories (Hötler et al. Citation2005; Niyuhire et al. Citation2007; Harloe et al. Citation2008). The interaction between stress, eCB system regulation, and memory processes may be complicated by the finding that homotypic and heterotypic stressor exposure results in contrasting changes in hippocampal AEA and 2-AG content (Hill et al. Citation2005, Citation2010b).
Stress-induced anxiety
Anxiety-like behavior in rodents is typically assessed in exploration- or avoidance-based tests, such as the elevated plus-maze or the light–dark chamber (Sousa et al. Citation2006). Other paradigms explicitly address approach behavior toward a potentially dangerous situation (e.g. Vogel conflict test; Sousa et al. Citation2006). Subsequently, stressor exposure is known to alter anxiety-like behavior. Interestingly, mutant mice lacking expression of CB1 receptors show increased anxiety-like behavior (e.g. decreased exploration of open arms on the elevated plus-maze, decreased social interaction, decreased exploration of brightly lit environments, and increased thigmotaxis) only under highly aversive conditions (i.e. bright light; Haller et al. Citation2004; Jacob et al. Citation2009). It is unknown to what extent these changes in anxiety-like behavior are mediated by alterations in HPA-axis activity (Steiner et al. Citation2008a). At least in the case of the elevated plus-maze, there is evidently an association between plasma corticosterone level and risk assessment (stretched/ attend posture and sniffing directed to open areas from enclosed arms of the maze; Rodgers and Dalvi Citation1997), but not the time spent on the open arms (Rodgers et al. Citation1999). CB1-deficient mice, which show increased HPA-axis activity (i.e. increased basal and stress-induced corticosterone and ACTH release; Steiner et al. Citation2008b), also display increased risk assessment on the elevated plus-maze (Jacob et al. Citation2009). However, this was not the case for conditional mutants, in which the CB1 receptor gene was specifically deleted from cortical glutamatergic neurons. In these mice, both HPA-axis activity and risk assessment behavior were unaltered, whereas social and novel object investigations were significantly reduced (Steiner et al. Citation2008b; Jacob et al. Citation2009).
A recent paper demonstrates that acute restraint stress leads to increased anxiety behavior on the elevated plus-maze 24 h later, independently of CB1 signaling within the ventral HPC (Campos et al. Citation2010). However, it also found that enhancement of eCB signaling via blockade of anandamide uptake attenuated stress-induced anxiety on the elevated plus-maze and reduced anxiety in the Vogel conflict test, if drug treatment was administered before testing. Remarkably, the same treatment promoted anxiety on the elevated plus-maze in unstressed rats. Another group found that treatment with a CB1 agonist increased anxiety in rats previously exposed to CMS even though the same treatment attenuated anxiety in the nonstressed group (Hill and Gorzalka Citation2004). There are evidently differential effects of cannabinoid treatment on anxiety-like behaviors in animals with and without previous stressor exposure. Prior stressor exposure may prime eCB signaling and/or alter the neuronal subpopulations activated by subsequent stressor exposure (Karst et al. Citation2010), which, in turn, could modify the effect of cannabinoid treatment on anxiety-related responses. In addition, differences in eCB signaling following acute vs. chronic stressor exposure may account for the divergent effects of cannabinoid treatment on anxiety seen in these two examples.
Rossi et al. (Citation2008) demonstrated in a number of elegant experiments that repeated social defeat stress (3–7 days) causes transient changes in eCB signaling within the dorsal striatum which revert within 3–5 days after the last defeat. These changes are characterized by a reduced sensitivity of GABAergic signaling to CB1 agonists in vitro as well as decreased exploration and increased anxiety in an open field (Rossi et al. Citation2008; De Chiara et al. Citation2010). These stress effects could be mimicked by corticosterone treatment and blocked by treatment with a GR antagonist, wheel running, or sucrose consumption (Rossi et al. Citation2008; De Chiara et al. Citation2010). Indeed, wheel running or sucrose consumption also enhances CB1-mediated control of inhibitory transmission in unstressed mice (De Chiara et al. Citation2010). Such functional changes in eCB signaling that occur following stress may relate to the previous contrasting effects of drug treatment on anxiety behaviors before and after stress exposure.
Stress and depression
The role of eCB system functioning in depression has been thoroughly considered in previous publications (Hill and Gorzalka Citation2005; Bambico and Gobbi Citation2008; Gorzalka et al. Citation2008; Hill et al. Citation2009a); however, we focus here on this relationship specifically in the context of stressor exposure. Anhedonia is a prominent symptom of major depression and is often measured as a decrease in sucrose preference in rodents. Restraint stress increases anhedonia in mice, which could be attenuated by treatment with CB1 receptor agonist or FAAH inhibitor, and intensified with CB1 antagonism (Rademacher and Hillard Citation2007). Interestingly, the effects of CB1 receptor blockade on sucrose preference increased with the number of restraint episodes, indicating an increased recruitment of eCB signaling with increased stressor exposure (Rademacher and Hillard Citation2007). Attenuation of anhedonia by treatment with an FAAH inhibitor has also been seen following CMS (Bortolato et al. Citation2007). In line with this, CMS leads to enhanced anhedonia in CB1 knockout vs. wild type mice (Martin et al. Citation2002). So far, results regarding eCB regulation of depression-like symptoms following chronic stress have been quite consistent, which indicates the therapeutic value of enhancing eCB signaling in the treatment of stress-related psychiatric conditions, such as depression. Vice versa, in clinical studies pharmacological blockade of CB1 led to a higher incidence of mood disturbances compared with placebo-treated controls (Christensen et al. Citation2007; Nissen et al. Citation2008), thus pointing to a preventive function of the eCB system in the development of depression and anxiety.
Perspectives
With such an abundance of data concerning the eCB system and stress, it becomes necessary to attempt to shape current predictions and guide future investigations with more comprehensive ideas and schemes. outlines a possible relationship between homeostatic and allostatic states, as well as how these states relate to habituation and recovery following stressor exposure. We will use this model to organize some of the previously discussed data and to highlight areas in need of further investigation. An allostatic state, as seen in , is defined as “altered and sustained activity levels of the primary mediators, e.g. glucocorticosteroids, that integrate physiology and associated behaviors in response to changing environments and challenges,” such as stress (McEwen and Wingfield Citation2003). Furthermore, such a state would be reached through increased allostatic load, being the “wear and tear on the body and brain resulting from chronic overactivity or inactivity of physiological systems” such as the HPA-axis in response to stress (McEwen Citation1998).
Figure 3. A schematic representation of the relationship between homeostatic and allostatic states. Allostatic load is the relative cost of the physiological responses to stress, such as HPA-axis activation (McEwen Citation1998; McEwen and Wingfield 2003). Prior to stressor exposure, homeostatic functioning is at a low allostatic load and eCB signaling helps maintain homeostasis by counteracting HPA-axis activation (Patel et al. Citation2004; Steiner and Wotjak Citation2008; Hill et al. Citation2009b; Ginsberg et al. Citation2010). During some stress protocols habituation-like processes may occur, which effectively reduce allostatic load and help reinstate homeostatic functioning. The eCB system evidently plays a key role in stress-induced habituation-like processes (Patel et al. Citation2005; Patel and Hillard Citation2008; Hill et al. Citation2010b). In cases where habituation does not occur, a relatively high incurred allostatic load may result in the development of an allostatic state. Several studies have investigated the role of eCB signaling in stress protocols in which habituation does not occur (Martin et al. Citation2002; Hill and Gorzalka Citation2004; Hill et al. Citation2005; Bortolato et al. 2007). Upon stressor termination, allostatic states may prevail leading to allostatic overload and conditions of chronic stress such as burnout, anxiety, and depression in humans. Evidence indicates that upregulation of eCB signaling may help prevent depression-like symptoms in animal models (Hill and Gorzalka Citation2005; Bambico and Gobbi Citation2008; Gorzalka et al. Citation2008; Hill et al. Citation2009a), but its role in the stress-induced development of these states is unclear. Recovery processes from allostatic states may occur to reduce allostatic load and reinstate homeostatic functioning. It has yet to be investigated what role eCB signaling may play in these recovery processes.
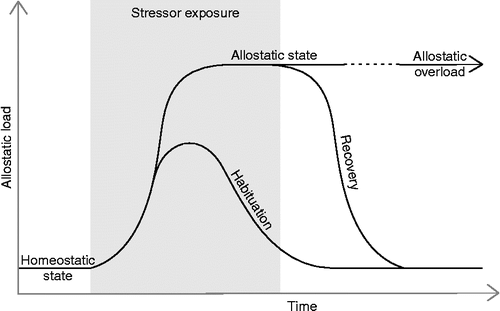
Upon stressor exposure, but depending on the stressor, as well as the organism involved, habituation-like processes may occur together with more complex associative learning processes (Grissom and Bhatnagar Citation2009), thereby protecting the organism from unnecessary stress responses and their detrimental effects. However, when habituation does not occur, the incurred allostatic load may advance an individual toward an allostatic state. Although such a state may be adaptive under certain circumstances (e.g. ongoing environmental threat), a sufficiently severe or prolonged allostatic state would lead to allostatic overload in which an organism is susceptible to the deleterious physiological and/or psychological effects of stress (McEwen and Wingfield Citation2003). Although the actual or relative allostatic load necessary for habituation vs. recovery processes to occur may differ from those illustrated in , the important defining feature is that habituation-like processes occur during stressor exposure and recovery processes occur following stressor termination. It is possible that an allostatic state is not reached if the stressor ends and recovery ensues before the state can develop. In Addition, an allostatic state may not develop during stressor exposure, but still develop after stress discontinuation, if recovery processes do not take place. Defining an allostatic state during stressor exposure is difficult; therefore, stress responses that endure (e.g. at least 24 h) after stressor termination would be of primary interest. Also, these prolonged allostatic states would closely relate to chronic stress-related psychological conditions in humans such as burnout, anxiety, and depression, in which therapeutic intervention is most likely.
Affirmation of habituation
The vast majority of investigations regarding the eCB system and stress are centered around homeostatic functioning, as deduced by basal measurement in contrast to those following acute stressor exposure, and habituation-like processes following homotypic stressor exposure. A representation of such research has been outlined in this paper and it is clear that the finer details of these processes have yet to be worked out, with close attention paid to the differing roles of AEA and 2-AG as well as variable functioning of the eCB system within distinct brain regions and neuronal populations. Nonetheless, one impediment in determining the exact functioning of the eCB system in stress adaptation is the question of whether, with a given stress protocol, habituation indeed occurs. It cannot be assumed that any repeated homotypic stressor protocol will result in habituation in stimulus-response pathways. This must, therefore, be confirmed and opposed to more complex associative learning mechanisms. We contend that the foremost evidence of stress habituation is a decline in HPA-axis functioning throughout a stress protocol, since it is overactivation of the HPA-axis that has been directly related to the deleterious effects of chronic stress (Selye Citation1950; Chrousos and Gold Citation1992; Bodnoff et al. Citation1995; Checkley Citation1996; de Kloet et al. Citation2005). Behavioral and biochemical data must be supplemented with analysis of HPA-axis functioning to evaluate whether habituation indeed takes place with a given stress protocol in a given species. Such studies have to explicitly consider the main criteria of habituation-like processes, including spontaneous recovery of the original HPA-axis response (Thompson and Spencer Citation1966; Grissom and Bhatnagar Citation2009). Importantly, with our current knowledge about the role of the eCB system (Patel et al. Citation2005; Kamprath et al. Citation2006; Patel and Hillard Citation2008), we have to regard habituation as an active (e.g. acute suppression of excitation by a primed eCB system) rather than a passive (e.g. deactivation of receptive structures) process.
Although changes in eCB signaling following repeated stress have been suggested to be involved in stress adaptation, some studies still label these changes as “impairments” without additional analysis of HPA-axis activity (Rademacher et al. 2007; Rossi et al. Citation2008; Hill et al. Citation2010b; Wamsteeker et al. Citation2010). Protocols that do not show signs of HPA-axis habituation may actually be better models for assessing eCB system mechanisms in an allostatic state.
Intensity, predictability, and controllability
Stressors can be described by a trinity of characteristics, namely, intensity, controllability, and predictability (Koolhaas, et al. Citation2011). Stressor duration is intrinsic to the concept of intensity as variations in stressor duration will accordingly affect stressor intensity. The eCB system plays a definitive role in stressor intensity as has been shown by increased recruitment of the system with increased stressor intensity/duration (Rademacher and Hillard Citation2007; Kamprath et al. Citation2009). Predictability and controllability also tend to co-vary, but may be distinguishable by unique physiological profiles before and after stressor exposure, respectively (Koolhaas et al. Citation2011). Controllability is inferred by the speed of recovery of the HPA-axis response following stressor exposure, and several lines of evidence have already linked eCBs with the resetting of the HPA-axis following stress (Di et al. Citation2003; Evanson et al. Citation2010; Koolhaas et al. Citation2011; Tasker and Herman Citation2011). Predictability, however, is the presence of an anticipatory response, such as an increase in corticosterone or noradrenaline secretion prior to an expected stressor (Koolhaas et al. Citation2011). Considering that between-session fear extinction occurs independently of CB1 receptor signaling, it is possible that eCBs would not contribute to stressor predictability; however, this is still an open question (Plendl and Wotjak Citation2010).
Acute stressor challenges give insight into eCB regulation of HPA-axis activity and may contribute to the long-term consequences of stressor exposure, but will do so in different ways from chronic stressor exposure. One of the reasons for this is that acute stressors do not contain any aspect of predictability or controllability, both of which depend on previous stressor exposure (Koolhaas et al. Citation2011). Investigations of homotypic stressor exposure can shed light on the relationship between eCB system functioning and controllability and predictability as well as their role in habituation and preventing the development of allostatic states and stress-related disease states. However, therapeutic intervention for chronic stress-related conditions, such as burnout, depression, or anxiety in humans, cannot be administered preventively, but only in response to the development of these conditions. Therefore, investigations must also focus on allostatic states and recovery processes.
Allostatic states and recovery processes
It still remains to be fully investigated what role the eCB system plays in chronic stress situations, in which habituation does not occur or does not suffice, namely in allostatic states and recovery. An understanding of these states can only be reached through the use of unpredictable and uncontrollable stressor exposure and, to date, only a few studies have assessed functioning of the eCB system following CMS, which have been outlined above. Although evidence indicates that enhanced eCB signaling may help attenuate depressive-like symptoms, there is not enough evidence to draw any general conclusions about eCB functioning during unpredictable, uncontrollable stressor exposure and the development of these depressive phenotypes. However, we would predict less eCB involvement during heterotypic stressor exposure in comparison with other chronic stress paradigms. Owing to the changing nature of the stressors, eCB-mediated habituation-like processes would be unable to develop and heterotypic stressors would, in essence, bypass eCB intervention, thus contributing to the development of allostatic states.
Once an allostatic state is reached, we propose that there are essentially two ways in which eCB system functioning may relate to this state and subsequent recovery processes. First, we consider the role of the system as a buffer against acute stress-induced increases in HPA-axis activity (Patel et al. Citation2004, Citation2005; Ganon-Elazar and Akirav 2009; Ginsberg et al. Citation2010). Through these mechanisms the eCB system may serve to counteract an allostatic state by providing a constant “pull” back toward homeostatsis and thereby promoting recovery. Second, some, or possibly all, allostatic states may be beyond the capabilities of the eCB system and could even infer a breakdown of effective functioning or even deterioration of the system. eCB-mediated habituation-like processes may not occur within all stress protocols simply because they lead to an allostatic load beyond the upper limits of the eCB system (defined e.g. by limited eCB synthesis and efficient uptake and degradation processes). In this case, an allostatic state may persist due, at least partially, to insufficient eCB functioning. This is in line with evidence indicating that chronic stress-related conditions, such as depression and cognitive decline, are linked to depreciated eCB system signaling (Martin et al. Citation2002; Hill and Gorzalka Citation2005; Hill et al. Citation2005, Citation2009a; Bambico and Gobbi Citation2008; Gorzalka et al. Citation2008). The boundary between functioning and nonfunctioning of the eCB system may occur at different degrees of allostatic load in various brain regions or neuronal networks and will likely be susceptible to individual differences. An especially informative method for determining the role of the eCB system in allostatic states and recovery processes is to compare the functioning of the system in animals that undergo recovery vs. those that do not, at a given time point following stressor termination.
Individual differences
The above leads us to another interesting aspect that has yet to be investigated, which is that of individual differences and how early life experiences affect the development of the eCB system and its stress-related functioning later in life. Multiple other parameters of could be influenced by individual differences that affect how an organism perceives and responds to a given stressor, for example, the conditions under which habituation-like processes or allostatic states occur or the timing and rate of recovery. Some studies have already indicated a relationship between early life stress and eCB system functioning in later life. For example, adult rats that received cannabinoid treatment in adolescence showed decreased depression-like behavior, which was reversed by early life stress exposure (Macrì and Laviola Citation2004; Marco et al. Citation2009). However, to our knowledge, there have been no investigations into how early life stressor exposure affects eCB system functioning during adulthood stressor exposure. Furthermore, individual differences in laboratory animals can arise without the experimental manipulation of early life stress and it should be noted that individual differences can be seen in stress responses even within genetically inbred species (Krishnan et al. Citation2007; Thoeringer et al. Citation2010). Individual variations in stressor-related responses and the related eCB system functioning can give valuable insight into possible therapeutic manipulations for stress-related conditions.
Conclusion
As demonstrated, the eCB system has been shown to play a role in a wide variety of stress-related processes. One common theme of eCB action is downregulation of excitatory (e.g. of glutamatergic) transmission, which prevents overexcitation in neuronal circuits responsible for hormonal and behavioral stress responses (). Another common and most important theme that has emerged is the importance of the interaction between eCB mechanisms and HPA-axis activity. Given their pronounced role in the functioning of the hormonal stress response, it is unsurprising that eCBs evidently closely interact with the HPA-axis in the context of most stress-related behaviors. However, ample contradictory data regarding these interactions still exist. Given that eCB signaling may have divergent properties within different brain structures and that AEA and 2-AG signaling may play quite distinct roles in relation to HPA-axis activity, it would be advisable to focus future behavioral research on specific brain areas or endogenous ligands. Moreover, administration of direct receptor agonists tells little about the role of the endogenous cannabinoid system in comparison with pharmacological enhancement of the endogenous cannabinoid signaling. Attention needs to be given to the relationship between the eCB system and stress responses involving the sympatho-adrenal medullary system, especially considering the extensive interactions between this system and the HPA-axis (Chrousos and Gold Citation1992).
We have attempted to organize some of our current knowledge about eCBs and stress within a scheme relating homeostatic and allostatic states. Although much research has focused on physiological functioning of the eCB system and its role in habituation, future investigations are advised to test the occurrence of habituation through measures of HPA-axis activity. Differences between homotypic and heterotypic chronic stressors relate to stressor intensity, predictability, and controllability, and eCBs may play a role in these processes. Important information regarding the therapeutic value of the eCB system in chronic stress-related conditions can be obtained through the study of allostatic states and recovery processes. Lastly, individual differences in the parameters of any of these processes or states may occur and could also provide information of therapeutic value.
Acknowledgments
This work has been supported by a Natural Sciences and Engineering Research Council of Canada Graduate Scholarship and a doctoral stipend from the Max Planck Society to C. J. R.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bambico FR, Gobbi G. 2008. The cannabinoid CB1 receptor and the endocannabinoid anadamide: Possible antidepressant targets. Expert Opin Ther Targets. 12:1347–1366.
- Bisogno T. 2008. Endogenous cannabinoids: Structure and metabolism. J Neuroendocrinol. 20 Suppl. 1: 1–9.
- Bitencourt RM, Pamplona FA, Takahashi RN. 2008. Facilitation of contextual fear memory extinction and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 18:849–859.
- Bodnoff SR, Humphreys AG, Lehman JC, Diamond DM, Rose GM, Meaney MJ. 1995. Enduring effects of chronic corticosterone treatment on spatial learning, synaptic plasticity, and hippocampal neuropathology in young and mid-aged rats. J Neurosci. 15:61–69.
- Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. 2007. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 62:1103–1110.
- Butler RK, Finn DP. 2009. Stress-induced analgesia. Prog Neurobiol. 88:184–202.
- Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. 2009. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci USA. 106:4888–4893.
- Campos AC, Ferreira FR, Guimaraes FS, Lemos JI. 2010. Facilitation of endocannabinoid effects in the ventral hippocampus modulates anxiety-like behaviors depending on previous stress experience. Neuroscience. 167:238–246.
- Cannon W. 1935. Stresses and strains of homeostasis. Am J Med Sci. 189:1–14.
- Checkley S. 1996. The neuroendocrinology of depression and chronic stress. Br Med Bull. 52:597–617.
- Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. 2005. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychophamacology. 30:516–524.
- Christensen R, Kristensen PK, Bartels EM, Biddal H, Astrup A. 2007. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet. 370:1706–1713.
- Chrousos GP, Gold PW. 1992. The concepts of stress and stress system disorders. J Am Med Assoc. 267:1244–1253.
- Coddington E, Christine L, Rose JD, Moore FL. 2007. Endocannabinoids mediate the effects of acute stress and corticosterone on sex behavior. Endocrinology. 148:493–500.
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. 1996. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 384:83–87.
- De Boer SF, Koopmans SJ, Slangen JL, van der Gugten J. 1990. Plasma catecholamine, corticosterone and glucose responses to repeated stress in rats: Effect of interstressor interval and length. Physiol Behav. 47:1117–1124.
- De Chiara V, Errico F, Musella A, Rossi S, Mataluni G, Sacchetti L, Siracusano A, Castelli M, Cavasinni F, Bernardi G, Usiello A, Centonze D. 2010. Voluntary exercise and sucrose consumption enhance cannabinoid CB1 receptor sensitivity in the striatum. Neuropsychopharmacology. 35:374–387.
- de Kloet RE, Joëls M, Holsboer F. 2005. Stress and the brain: From adaptation to disease. Nat Rev Neurosci. 6:463–475.
- de Oliveira Alvares L, Engelke DS, Diehl F, Scheffer-Teixeira R, Haubrich J, Cassini L, Molina VA, Quillfeldt JA. 2010. Stress response recruits the hippocampal endocannabinoid system for the modulation of fear memory. Learn Mem. 17:202–209.
- Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. 1988. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 34:604–613.
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. 1992. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 258:1946–1949.
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. 2003. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: A fast feedback mechanism. J Neurosci. 23:4850–4857.
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J, Piomelli D. 1994. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 372:686–691.
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. 2002. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 99:10819–10824.
- Egertová M, Giang DK, Cravatt BF, Elphick MR. 1998. A new perspective on cannabinoid signaling: Complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 265:2081–2085.
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. 2010. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 151:1–9.
- Finn DP. 2010. Endocannabinoid-mediated modulation of stress responses: Physiological and pathophysiological significance. Immunobiology. 215:629–646.
- Finn DP, Beckett SRG, Richardson D, Kendall DA, Marsden CA, Chapman V. 2004. Evidence for differential modulation of conditioned aversion and fear-conditioned analgesia by CB1 receptors. Eur J Neurosci. 20:848–852.
- Ford GK, Finn DP. 2008. Clinical correlates of stress-induced analgesia: Evidence from pharmacological studies. Pain. 140:3–7.
- Ganon-Elazar E, Akirav I. 2009. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 29:11078–11088.
- Ginsberg AB, Pecoraro NC, Warne JP, Horneman HF, Dallman MF. 2010. Rapid alterations of stress-induced hypothalamic–pituitary–adrenal hormone secretion in the rat: A comparison of glucocorticoids and cannabinoids. Stress. 13:248–257.
- Goani Y, Mechoulam R. 1964. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc. 86:1646–1647.
- Goparaju SK, Ueda N, Taniguchi K, Yamamoto S. 1999. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol. 57:417–423.
- Gorzalka BB, Hill MN, Hillard CJ. 2008. Regulation of endocannabinoid signaling by stress: Implications for stress-related affective disorders. Neurosci Biobehav Rev. 32:1152–1160.
- Grissom N, Bhatnagar S. 2009. Habituation to repeated stress: Get used to it. Neurobiol Learn Mem. 92:215–224.
- Haller J, Varga B, Ledent C, Barna I, Freund TF. 2004. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 19:1906–1912.
- Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R. 2001. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci USA. 98:3662–3665.
- Harloe JP, Thorpe AJ, Lichtman AH. 2008. Differential endocannabinoid regulation of extinction in appetitive and aversive Barnes maze tasks. Learn Mem. 15:806–809.
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. 1990. Cannabinoid receptor localization in the brain. Proc Natl Acad Sci USA. 87:1932–1936.
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. 1991. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J Neurosci. 11:563–583.
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. 2003. Central mechanisms of stress integration: Hierarchical circuitry controlling hypothalamo–pituitary–adrenocortical responsiveness. Front Neuroendocrinol. 24:151–180.
- Herman JP, Ostrander MM, Mueller NK, Figueiredo H. 2005. Limbic system mechanisms of stress regulation: Hypothalamo–pituitary–adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 29:1201–1213.
- Hill MN, Gorzalka BB. 2004. Enhancement of anxiety-like responsiveness to the cannabinoid receptor agonist HU-210 following chronic stress. Eur J Pharmacol. 499:291–295.
- Hill MN, Gorzalka BB. 2005. Is there a role for the endocannabinoid system in the etiology and treatment of melancholic depression?. Behav Pharmacol. 16:333–352.
- Hill MN, McEwen BS. 2009. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci USA. 106:4579–4580.
- Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, Gorzalka BB. 2005. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 30:508–515.
- Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G. The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci. 2009a; 30:484–493.
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic–pituitary–adrenal axis. Neuropsychopharmacology. 2009b; 34:2733–2745.
- Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010a; 35:1333–1338.
- Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TTY, Gray JM, Hillard CJ, Gorzalka BB, Viau V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci USA. 2010b; 107:9406–9411.
- Hillard CJ, Harris RA, Bloom AS. 1985. Effects of cannabinoids on physical properties of brain membranes and phospholipid vesicles: Fluorescence studies. J Pharmacol Exp Ther. 232:579–588.
- Hinz B, Hirschelmann R. 2000. Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharm Res. 17:1273–1277.
- Hohmann AG, Suplita RL. 2006. Endocannabinoid mechanisms of pain modulation. AAPS J. 8:E693–E708.
- Hölter SM, Kallnik M, Wurst W, Marsicano G, Lutz B, Wotjak CT. 2005. Cannabinoid CB1 receptor is dispensable for memory extinction in an appetitively-motivated learning task. 2005. Eur J Pharmacol. 7:69–74.
- Howlett AC. 1987. Cannabinoid inhibition of adenylate cyclase: Relative activity of constituents and metabolites of marihuana. Neuropharmacology. 26:507–512.
- Howlett AC, Fleming RM. 1984. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 26:532–538.
- Jacob W, Yassouridis A, Marsicano G, Monory K, Lutz B, Wotjak CT. 2009. Endocannabinoids render exploratory behaviour largely independent of the test aversiveness: Role of glutamatergic transmission. Genes Brain Behav. 8:685–698.
- Kamprath K, Wotjak CT. 2004. Nonassociative learning processes determine expression and extinction of conditioned fear in mice. Learn Mem. 11:770–786.
- Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, Lutz B, Wotjak CT. 2006. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 26:6677–6686.
- Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, Wotjak CT. 2009. Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signaling. Genes Brain Behav. 8:203–211.
- Kamprath K, Romo-Parra H, Häring M, Gaburro S, Doengi M, Lutz B, Pape H. 2010. Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology. 36:652–663.
- Kant GJ, Eggleston T, Landman-Roberts L, Kenion CC, Driver GC, Meyerhoff JL. 1985. Habituation to repeated stress is stressor specific. Pharmacol Biochem Behav. 22:631–634.
- Karst H, Berger S, Erdmann G, Schütz G, Joëls M. 2010. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci USA. 107:14449–14454.
- Keller-Wood ME, Dallman MF. 1984. Corticosterone inhibition of ACTH secretion. Endocr Rev. 5:1–24.
- Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flügge G, Korte SM, Meerlo P, Murison R, Oliver B, Palanza P, Richter-Levin G, Sgoifo A, Steimer T, Stiedl O, van Dijk G, Wöhr M, Fuchs E. 2011. Stress revisited: A critical evaluation of the stress concept. Neurosci Biobehav Rev. 35:1291–1301.
- Krishnan V, Han M, Graham DL, Berton O, Renthal W, Russo SJ, LaPlant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. 2007. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 131:391–404.
- Lazarus RS. 1966. Psychological stress and the coping process. New York: McGraw-Hill.
- Lazarus RS, Launier R. 1978. Stress-related transactions between person and environment. In: Pervin LA, Lewis M. editors. Perspectives in interactional psychology. New York: Plenum287–327.
- Lazarus RS, Folkman S. 1984. Stress, appraisal, and coping. New York: Springer.
- Leggett JD, Aspley S, Beckett SRG, D'Atona AM, Kendall DA, Kendall DA. 2004. Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br J Pharmacol. 141:253–262.
- Lovinger DM. 2007. Endocannabinoid liberation from neurons in transsynaptic signaling. J Mol Neurosci. 33:87–93.
- Mackie K, Hille B. 1992. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 89:3825–3829.
- Mackie K, Lai Y, Westenbroek R, Mitchell R. 1995. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 15:6552–6561.
- Macrì S, Laviola G. 2004. Single episode of maternal deprivation and adult depressive profile in mice: Interaction with cannabinoid exposure during adolescence. Behav Brain Res. 154:231–238.
- Malcher-Lopes R, Di S, Marcheselli VS, Weng F, Stuart CT, Bazan NG, Tasker JG. 2006. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 26:6643–6650.
- Marco EM, Adriani A, Llorente R, Laviola G, Viveros M. 2009. Detrimental psychophysiological effects of early maternal deprivation in adolescent and adult rodents: Altered responses to cannabinoid exposure. Neurosci Biobehav Rev. 33:498–507.
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. 2002. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 418:530–534.
- Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O. 2002. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology. 159:379–387.
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. 1990. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 346:561–564.
- McEwen BS. 1998. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 840:33–44.
- McEwen BS, Wingfield JC. 2003. The concept of allostasis in biology and biomedicine. Horm Behav. 43:2–15.
- Mikics É, Dombi T, Barsvári B, Varga B, Ledent C, Freund TF, Haller J. 2006. The effects of cannabinoids on contextual conditioned fear in CB1 knockout and CD1 mice. Behav Pharmacol. 17:223–230.
- Moreira FA, Wotjak CT. 2010. Cannabinoids and anxiety. Curr Top Behav Neurosci. 2:429–450.
- Munro S, Thomas KL, Abu-Shaar M. 1993. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 365:61–65.
- Natelson BH, Ottenweller JE, Cook JA, Pitman D, McCarty R, Tapp WN. 1988. Effect of stressor intensity on habituation of the adrenocortical stress response. Physiol Behav. 43:41–46.
- Nissen SE, Nicholls SJ, Wolski K, Rodés-Cabau J, Cannon CP, Deanfield JE, Després JP, Kastelein JJ, Steinhubl SR, Kapadia S, Yasin M, Ruzyllo A, Gaudin C, Job B, Hu B, Bhatt DL, Lincoff AM, Tuzcu EM, STRADIVARIUS Investigators. 2008. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: The STRADIVARIUS randomized controlled trial. J Am Med Assoc. 299:1547–1560.
- Niyuhire F, Varvel SA, Thorpe AJ, Stokes RJ, Wiley JL, Lichtman AH. 2007. The disruptive effects of the CB1 receptor antagonist rimonabant on extinction learning in mice are task-specific. Psychopharmacology. 191:223–231.
- Pamplona FA, Bitencourt RM, Takahashi RN. 2008. Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol Learn Mem. 90:290–293.
- Patel S, Hillard CJ. 2008. Adaptations in endocannabinoid signaling in response to repeated homotypic stress: A novel mechanism for stress habituation. Eur J Neurosci. 27:2821–2829.
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. 2004. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic–pituitary–adrenal axis. Endocrinology. 145:5431–5438.
- Patel S, Roelke CT, Rademacher DJ, Hillard CJ. 2005. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signaling. Eur J Neurosci. 21:1057–1069.
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. 2009. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuroppsychopharmacology. 34:2699–2709.
- Pecoraro N, Dallman MF, Warne JP, Ginsberg AB, Laugero KD, la Fleur SE, Houshyar H, Gomez F, Bhargava A, Akana SF. 2006. From Malthus to motive: How the HPA axis engineers the phenotype, yoking needs to wants. Prog Neurobiol. 79:247–340.
- Piomelli D. 2003. The molecular logic of endocannabinoid signaling. Nat Rev Neurosci. 4:873–884.
- Plendl W, Wotjak CT. 2010. Dissociation of within- and between-session extinction of conditioned fear. J Neurosci. 30:4990–4998.
- Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC. 2002. Characterization of a novel endocannabinoid, Virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther. 301:1020–1024.
- Rademacher DJ, Hillard CJ. 2007. Interactions between endocannbinoids and stress-induced decreased sensitivity to natural reward. Prog Neuropsychopharmacol Biol Psychiatry. 31:633–641.
- Rademacher DJ, Meier SE, Shi L, Ho WSV, Jarrahian A, Hillard CJ. 2008. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 54:108–116.
- Rodgers RJ, Dalvi A. 1997. Anxiety, defence and the elevated plus-maze. Neurosci Biobehav Rev. 21:801–810.
- Rodgers RJ, Haller J, Holmes A, Halasz J, Walton TJ, Brain PF. 1999. Corticosterone response to the plus-maze: High correlation with risk assessment in rats and mice. Physiol Behav. 68:47–53.
- Rossi S, De Chiara V, Musella A, Kusayanagi H, Mataluni G, Bernardi G, Usiello A, Centonze D. 2008. Chronic psychoemotional stress impairs cannabinoid-receptor-mediated control of GABA transmission in the striatum. J Neurosci. 28:7284–7292.
- Selye H. 1950. Stress and the general adaptation syndrome. Br Med J. 4667:1383–1392.
- Sousa N, Almeida OF, Wotjak CT. 2006. A hitchhiker's guide to behavioral analysis in laboratory rodents. Genes Brain Behav. 5 Suppl. 2: 5–24.
- Steiner MA, Wotjak CT. 2008. Role of the endocannabinoid system in regulation of the hypothalamic–pituitary–adrenocortical axis. Prog Brain Res. 170:397–432.
- Steiner MA, Marsicano G, Nestler EJ, Holsboer F, Lutz B, Wotjak CT. Antidepressant-like behavioral effects of impaired cannabinoid receptor type 1 signaling coincide with exaggerated corticosterone secretion in mice. Psychoneuroendocrinology. 2008a; 33:54–67.
- Steiner MA, Marsicano G, Wotjak CT, Lutz B. Conditional cannabinoid receptor type 1 mutants reveal neuron subpopulation-specific effects on behavioral and neuroendocrine stress responses. Psychoneuroendocrinology. 2008b; 33:1165–1170.
- Stella N, Schweitzer P, Piomelli D. 1997. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 388:773–778.
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 1995. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 215:89–97.
- Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the HPA axis. Stress. 2011 in press.
- Tasker JG, Di S, Malcher-Lopes R. 2006. Minireview: Rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 147:5549–5556.
- Thoeringer CK, Pfeiffer UJ, Rammes G, Pamplona FA, Moosmang S, Wotjak CT. 2010. Early life environment determines the development of adult phobic-like fear responses in BALB/cAnN mice. Genes Brain Behav. 9:947–957.
- Thompson RF, Spencer WA. 1966. Habituation: A model phenomenon for the study of neuronal substrates of behaviour. Psychol Rev. 73:16–43.
- Tsigos C, Chrousos GP. 2002. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 53:865–871.
- Twitchell W, Brown S, Mackie K. 1997. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 78:43–50.
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. 2005. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 310:329–332.
- Wamsteeker JI, Kuzmiski JB, Bains JS. 2010. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 30:1118–11196.
- Willner P. 1997. Validity, reliability and utility of the chronic mild stress model of depression: A 10-year review and evaluation. Psychopharmacology. 134:319–329.
- Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. 1987. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by tricyclic antidepressant. Psychopharmacology. 93:358–364.