Abstract
Stress during early life can impact the developing brain and increase vulnerability to mood disorders later in life. Here, we argue that epigenetic mechanisms can mediate the gene–environment dialogue in early life and give rise to persistent epigenetic programming of adult physiology eventually resulting in disease. Early life stress in mice leads to epigenetic marking of the arginine vasopressin (AVP) gene underpinning sustained expression and increased hypothalamic–pituitary–adrenal axis activity. This epigenetic memory is laid down in the parvocellular neurons of the paraventricular nucleus and involves Ca2+/calmodulin kinase-mediated phosphorylation of the methyl-CpG binding domain protein MeCP2 leading to dissociation from its DNA-binding site and derepression of the AVP gene. The reduced occupancy of MeCP2 during this early stage of life facilitates the development of hypomethylation at the AVP enhancer, which sustains derepression throughout later life and thereby serves to hardwire early life experiences. The sequential order of these events may represent a critical time window for the preventive therapy of severe trauma.
Early life decides
The philosopher, Jan-Paul Sartre, proclaimed that all of psychology can be explained with two simple words—childhood decides (Satre Citation1964). Exposure to various adverse experiences during early life is a well-documented risk factor for psychopathology. In humans, a childhood history of abuse, trauma or neglect is well known to increase later susceptibility to affective disorders (Heim and Nemeroff Citation2001; Heim and Nemeroff Citation2002; Lupien et al. Citation2009; Green et al. Citation2010). The fact that stressors during early life associate with elevated risks for affective disorders developing not only in childhood but also in adulthood suggests that such exposures during the early periods of life confer an increased risk to psychopathology that persists throughout later life. One explanation for this increased risk for mental diseases is that the experience of stress in early life influences the development of stress-response systems.
What then are possible biological underpinnings by which early life experiences are able to shape the brain systems underlying emotion and behaviour? Developmental plasticity defines an organism's ability to adapt to the environment during early life and to implement long-lasting changes in physiology (for review, see Gluckman et al. Citation2008; Hochberg et al. Citation2011). Activity-dependent plasticity allows for an interaction of environmental signals (Katz and Shatz Citation1996) with the genetic blueprint to mould developmental trajectories in the central nervous system governing perception and subsequent responses to the environment (). Although organisms are likely to adapt to their environment throughout life, the most critical stages are represented by the prenatal period continuing through the neonatal and early adolescent stages in which the environment is able to fine-tune neural circuitry through various mechanisms (for review, see Heim et al. Citation2004).
Figure 1. Long-term programming of the stress system takes place during critical developmental time windows. The environment during early life can persistently alter the expression levels of key genes by epigenetic marking thus initiating adjustments in behaviour, neuroendocrine and stress responsivity throughout later life. The nature of the environment and experiences throughout later life, in addition to the impact of biological processes associated with aging and genetic sex, may exacerbate the programming established during early life resulting in increased vulnerability to and manifestation of mood disorders.
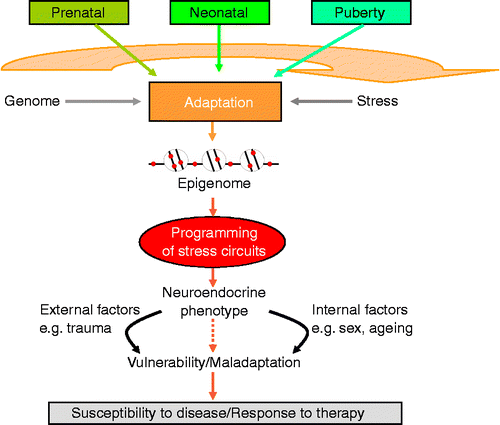
As noted earlier, during critical periods of prenatal and postnatal mammalian development, stress can influence developmental pathways thereby inducing permanent structural and regulatory changes predisposing to stress-related diseases in later life. Core to the long-term regulation of systems controlling stress responsivity is the hypothalamic–pituitary–adrenal (HPA) axis. Activation of this unit by exposure to stress triggers the release of corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP) from the paraventricular nucleus (PVN) of the hypothalamus. These neuropeptides bind to their receptors at the anterior pituitary to stimulate synthesis and release of adrenocorticotrophic hormone (ACTH) that then acts on the adrenal cortex to stimulate production and secretion of the stress steroids cortisol (primates) or corticosterone (rodents). Once homeostatic demands are met, negative feedback loops, provided by glucocorticoid receptor (GR) and mineralocorticoid receptor, attenuate HPA axis responsiveness and reinstate previous or adjusted ground states (De Kloet et al. Citation1998).
Effects of early life adversity
Substantial evidence supports the existence of long-term effects on HPA axis functioning following early life stress; these effects persist into adulthood and are accompanied by lasting behavioural changes. In clinical studies, early life stress has been shown to be a strong predictor of ACTH responsiveness (Heim et al. Citation2002) and impaired inhibitory feedback regulation of the HPA axis (Heim et al. Citation2008) as deduced by a dexamethasone/CRH challenge tests. Rodent studies have broadly demonstrated profound long-term effects of adverse conditions during early life on HPA functioning, including changes associated with increased glucocorticoid response to subsequent stressors, increased plasma levels of ACTH, reduced GR expression in the hippocampus, and changes in CRH and AVP in the hypothalamus (for review, see McCormick et al. Citation2010).
In view of persuasive evidence from clinical and animal studies regarding the intimate link between stress and HPA functioning, a strong case can be made that early life stress may lead to enduring dysregulation of the HPA axis and predispose to manifest psychiatric disorders in later life.
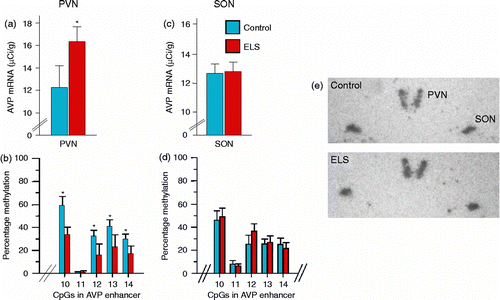
To elucidate the molecular underpinnings that allow early life adversity to programme lifelong health and behaviour, we have used periodic postnatal infant–mother separation in mice (Murgatroyd et al. Citation2009) as a paradigm for inducing early life stress (for review, see Pryce and Feldon Citation2003). This maternal separation paradigm, involving the separation of pups from their mother for 3 h each day during the first 10 days of life, induced lifelong elevated corticosterone secretion, heightened endocrine responsiveness to subsequent stressors and altered feedback inhibition of the HPA axis. In addition, early life stress-treated mice showed reduced stress-coping ability and memory deficits. Interestingly, AVP expression was persistently increased in the parvocellular subpopulation of paraventricular neurons but not in the supraoptic nucleus (SON), whereas CRH expression remained unaltered (). This elevation starts at 10 days of life, directly following the period of maternal separation, and lasts for at least 1 year. Treatment of the early life stressed mice with an AVP V1b receptor antagonist normalized the increased stress responses, indicating a central role for the increased AVP expression in the phenotype. In contrast to HPA axis over-activity, serum osmolality, controlled by AVP from the SON, was not altered by the early life stress, again attesting to the tissue- and neurocircuitry-specific nature of the early life stress-induced change in gene expression. AVP critically modulates mood behaviours (Landgraf Citation2006; Goekoop et al. Citation2010), in addition to playing an important role in the postnatal development and functional maturation of the pituitary–adrenal axis; in particular, AVP potentiates the actions of CRH under circumstances demanding sustained activation of the pituitary and adrenal glands (Aguilera and Rabadan-Diehl Citation2000; Engelmann et al. Citation2004).
Figure 2. Early life stress induces tissue-specific programming of AVP. Early-life stress leads to (a) increased AVP mRNA expression and (b) hypomethylation of the AVP enhancer in the PVN. (c) AVP mRNA expression and (d) DNA methylation were unaltered in the SON. Representative in situ hybridizations of AVP expression in the hypothalamus of control and early life stressed mice (e).
The epigenome and early life adversity
These findings raised the questions by which mechanisms AVP gene expression becomes tissue-specifically programmed and how they couple to the experience of an adverse early life environment.
During development, the genome is read out to express appropriate sets of genes in specific tissues at given time points. Sustained gene expression is accomplished by epigenetic mechanisms that govern accessibility of the DNA to the machinery driving gene expression. Modification of histones that package the DNA and methylation of cytosines in cytosine–guanine (CpG) dinucleotides represents the best-understood epigenetic marks (Allis et al. Citation2007; Eccleston et al. Citation2007). In contrast to the DNA sequence that is identical in all tissues, the patterns of epigenetic marks (i.e. epigenomes) are tissue-specific. Therefore, a genome can be considered to contain two layers of information: the DNA sequence inherited from our parents which is fixed throughout life and mostly identical in all cells and tissues of our body, and chromatin and DNA methylation patterns which are cell- and tissue-specific.
Apart from controlling constitutive gene expression, epigenetic mechanisms can also serve to fine-tune gene expression potential in response to environmental cues, thus conferring an additional level of plasticity to the hard-coded genome. Among these, DNA methylation has been the most studied with regard to understanding early life experiences and their neurobiological sequelae. The stable nature of DNA methylation renders it an ideal template for underpinning sustained gene effects controlling brain function and behaviour from early development to adulthood.
Studies from Meaney and coworkers (Weaver et al. Citation2004) have shown that rat offspring receiving poor maternal care develop behavioural alterations associated with reduced GR expression in the hippocampus resulting in an attenuated negative feedback regulation of the stress axis. The altered transcriptional programming of the GR occurred through increased DNA methylation at a specific site in a promoter of one of the alternative untranslated first exons of the gene. Interestingly, the DNA methylation changes developed rapidly in early life became fixed and persisted into adulthood suggesting that during early periods of life, DNA methylation patterns can be strongly impacted by the environment. Strikingly, suicide victims with a history of childhood abuse were also found to have decreased GR expression and increased methylation in the hippocampus at the analogous site in the human GR gene (McGowan et al. Citation2009). Ribosomal genes have likewise been reported to show altered DNA methylation in hippocampal tissues of suicide victims with a history of abuse and neglect (McGowan et al. Citation2008). Thus, it has been proposed that conditions of early life environment can evoke changes in DNA methylation facilitating epigenetic programming of critical genes involved in regulating stress responsivity that may in turn manifest with neuroendocrine and behavioural symptoms in adulthood (McGowan et al. Citation2009; Murgatroyd et al. Citation2010a) ().
To test whether experience-coupled changes in DNA methylation may explain the sustained AVP expression in our mouse model of maternal separation, we bisulphite sequenced the regions of the AVP locus to determine DNA methylation patterns. When comparing these epigenetic patterns between early life stressed and control mice, we discovered a long-lasting hypomethylation of CpG residues at a key regulatory region of the AVP gene. These changes were confined to the PVN of the hypothalamus and were absent in the SON; in other words, they occurred only in those cells showing altered AVP expression (). The early life stress-induced DNA hypomethylation further centred to a region of the downstream AVP enhancer containing high-affinity context-dependent DNA-binding sites for MeCP2 (Murgatroyd et al. Citation2009) conforming to sequence constraints previously described as important for the binding of this protein (Klose et al. Citation2005).
DNA methylation is interpreted by a family of methyl-CpG binding domain (MBD) containing proteins comprising the aforementioned MeCP2, MBD1, MBD2, MBD3 and MBD4; the first three of which couple DNA methylation to transcriptional repression (Klose and Bird Citation2006; Allis et al. Citation2007). Using chromatin immunoprecipitation (ChIP) experiments to determine levels of MBD-binding proteins, we found that early life stress responsive CpG residues were targeted specifically by MeCP2 and were spared from occupancy by the other MBD proteins (Murgatroyd et al. Citation2009).
As mentioned, the increased expression of AVP becomes apparent in postnatal day 10 mice, directly after the final maternal separation procedure, suggesting that the neuropeptide is acutely responsive to stress during this early life period (Veenema et al. Citation2006). Using in vivo ChIP experiments (CitationMurgatroyd et al. 2011), we found that MeCP2 occupancy at the AVP enhancer in the PVN was strongly reduced during this early period in stressed mice when compared with that in controls (Murgatroyd et al. Citation2009). This was unexpected as analysis of DNA methylation at the AVP enhancer revealed no differences between control and early life stressed mice at this stage, indicating that the upregulation of AVP during early life does not directly associate with changes in DNA methylation. Instead, it would appear that MeCP2 binding to this region is not primarily governed in cis at the level of the DNA.
What then are the signals controlling MeCP2 occupancy at this early step? Experience-driven synaptic activity causes membrane depolarization and calcium influx into select neurons, which in turn induce a wide variety of cellular responses that are critical for the development of new circuits and alter synaptic connectivity within existing ones (Greer and Greenberg Citation2008). Many of the signalling pathways that link neuronal activity to transcription and the regulation of complex programmes of gene expression have been identified in the last years (Flavell and Greenberg Citation2008). Among these, neuronal depolarization has also been shown to trigger Ca2+-dependent phosphorylation of MeCP2, causing dissociation of MeCP2 from the Bdnf promoter and derepression (Chen et al. Citation2003; Martinowich et al. Citation2003). Recent evidence further suggests that some drugs may function through pathways involving phosphorylation of MeCP2 in the brain (Deng et al. Citation2010).
Phosphorylation of rat MeCP2 at serine 421 has been demonstrated to be mediated through the de novo activity of the serine/threonine kinase, Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Zhou et al. Citation2006). In accord, we discovered that phosphorylation of the homologous serine residue 438 in the mouse MeCP2 leads to dissociation from the AVP enhancer in a hypothalamic cell line. Because MeCP2 occupancy at the AVP locus appears to be regulated by depolarization-dependent Ca2+/CaMKII-mediated pathways, we further tested whether the maternal separation stress stimulus may likewise lead to MeCP2 phosphorylation in the PVN. Indeed, we detected increased MeCP2 S438 phospho-immunoreactivity in the parvocellular AVP-expressing neurons of 10-day-old early life stressed mice. In addition, these neurons also displayed increases in phosphorylated CaMKII (the enzymatically active form of CaMKII) immunoreactivity. These findings are compatible with the hypothesis that CaMKII might mediate activity-dependent MeCP2 phosphorylation (Zhou et al. Citation2006). Apart from CaMKII, other calmodulin kinase members, such as CaMKIV (Tao et al. Citation2009), or unrelated phosphokinases (Bracaglia et al. Citation2009), could target additional sites on the MeCP2 protein (Tao et al. Citation2009). These differences in phosphorylated MeCP2 and CaMKII were specific to the parvocellular neurons of the PVN, whereas the SON showed no such changes in response to early life stress (Murgatroyd et al. Citation2009). Together, these findings support the concept that early life stress leads to activation of specific neuronal circuits controlling HPA axis activity via depolarization of paraventricular neurons, which in turn couples to MeCP2-S438 phosphorylation, and ultimately, relief of its occupancy at the AVP enhancer.
Maintenance of epigenetic marks
We then tested for MeCP2 and CaMKII phosphorylation in the PVN of adult (6-week old) early life stress and control mice and found that, even though AVP peptide and mRNA expression were persistently upregulated, differences in phosphorylated CaMKII and MeCP2 immunoreactivity declined and were barely distinguishable between adult early life stressed and control mice. At the same age, however, DNA hypomethylation had gradually evolved in early life stressed mice and underpinned reduced MeCP2 occupancy at the AVP enhancer (Murgatroyd et al. Citation2009). This order of events suggests that MeCP2 maintains AVP repression by the recruitment of DNA methyltransferases (DNMTs) to protect DNA methylation in the long run—a scenario supported by our sequential ChIP experiments showing a colocalization of MeCP2 with DNMT3a and DNMT3b (CitationMurgatroyd and Spengler 2011). We, therefore propose that early life stress contrarily leads to the phosphorylation and dissociation of MeCP2 at the AVP enhancer in turn paving erosion of DNA methylation and further reinforcing the loss of MeCP2 binding.
Interestingly, in the control mice, an age-associated demethylation also occurs though, critically, the region of the enhancer marked by early life stress is spared, supporting the importance of this region in AVP regulation (Murgatroyd et al. Citation2010b). As PVN neurons are postmitotic, such reductions in methylation would suggest the presence of active demethylation, though the identity of any ‘DNA demethylase’, and what roles such mechanisms may have during ageing remain elusive (Meaney and Ferguson-Smith Citation2010).
Establishment of epigenetic marks
Contrary to 6-week, 3-month and 1-year-old mice, the mice directly after maternal separation at 10 days of age show AVP repression irrespective of changes in DNA methylation. Although this may in some part reflect altered transcription factor-regulated activity of AVP at this stage directly following the stressor, it also highlights the fact that the reduced MeCP2 occupancy at the AVP enhancer, we see in our ChIP experiments, occurs irrespective of changes in DNA methylation. This leads us to propose that reduced MeCP2 occupancy facilitates increased active chromatin marks and relief of AVP repression irrespective of changes in DNA methylation. Indeed, sequential in vivo ChIP experiments show that MeCP2 preferentially associates with the repressive dimethylated histone H3K9 over the active acetylated H3 histone mark at the AVP locus (CitationMurgatroyd and Spengler 2011) and further colocalizes with histone deacetylation (HDAC) (CitationMurgatroyd and Spengler 2011; Uchida et al. Citation2011), supporting previous evidence for a role of MeCP2 as an epigenetic platform upon which HDAC, H3K9 methylation and DNA methylation are carried out to confer transcriptional repression and gene silencing () (Klose and Bird Citation2006; Chahrour and Zoghbi Citation2007; Murgatroyd et al. Citation2009; Murgatroyd et al. Citation2010a). Whether such chromatin marks already associate with MeCP2 binding in the postnatal day 10 mice needs to be tested as does the sequential order of epigenetic components, which establish DNA methylation at this region. Certainly, gaining insight into the origin of an epigenetic print at the AVP locus will provide us with a clearer picture on its regulation during critical time points and the players involved.
Figure 3. Epigenetic programming of AVP. Experience-dependent activation of neurons in the PVN during early life leads to phosphorylation of MeCP2 inhibiting its DNA binding to and repression of AVP. In the absence of MeCP2-binding DNA, hypomethylation gradually evolves in early life stressed mice underpinning reduced MeCP2 occupancy at the AVP enhancer and maintaining epigenetic control of AVP expression into later life.
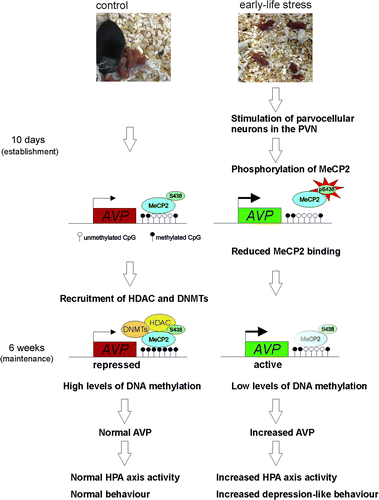
Taken together, the initial stimulus of early life stress initiates an uncoupling of MeCP2 occupancy that subsequently leads to the hard-coding of the early life experience at the level of DNA methylation (). In consequence, these mice are permanently caught by their adverse early life experience and their latent scars become easily unmasked, as evidenced by altered endocrine responses to further stress exposures later in life.
Time windows for therapeutic interventions
Epigenome-wide analyses using high-throughput sequencing are now moving research on from single-gene analyses to the analysis of all the epigenetic marks throughout a genome. Considering the evidence that epigenetic modifications are sensitive to changes in the environment, it is anticipated that such sequencing might identify epigenomic signatures for the basic molecular dysregulations underlying pathological conditions such as mental disorders. With the recent establishment of the International Epigenome Consortium, to address the possibility of generating epigenomes of various tissues, it is hoped that such approaches might offer the potential of epigenetic references for ‘diseased’ tissues (Albert Citation2010). Although such a strategy appears highly attractive in the field of cancer research addressing clonally expanded cell populations, rather than heterogenous tissues, the field of epigenetic association studies in mental illnesses might be more challenging. Epigenetic modifications found to date in animal models and postmortem brain tissues are seldom ‘all or none’ but gradual, and seem to occur in a highly cell-type manner. In this view, the extraordinary complexity and heterogeneity of neural tissues pose a major hurdle in deriving epigenetic biomarkers in psychiatric disease.
Ultimately, the identification of ‘epigenetic biomarkers’ in distinct genomic regions may provide important information for the understanding of biological processes underlying mental diseases and thus allow for the development and design of new therapeutics. The tissue-specific nature of epigenetic changes would demand the testing of human postmortem brain material, and unless parallel changes occur in the blood, or other material available for biopsy, the prospect of diagnostic epigenetic testing for mental diseases would appear limited.
However, the use of such data could be invaluable for allowing greater insight into how epigenetic marks are established in a temporal and gene-specific manner and in understanding how experience-dependent epigenetic marks may undergo the transition from a preliminary, labile state to a hard-coded, stable state shedding new light on mechanisms controlling epigenetic programming. Indeed, in our studies, epigenetic marking of the methylation landmarks in the AVP enhancer persisted under vasopressin receptor blockade in adult (3 months) mice consistent with the concept that the early life stress had already engraved a lasting cellular memory (CitationMurgatroyd and Spengler 2011). The question, however, remains of whether critical windows for timely psychotherapeutic and pharmacological interventions following exposure to severe trauma may exist at periods prior to DNA methylation-encoded maturation of epigenetic marks. In this view, different environmental manipulations were reported to attenuate HPA and behavioural responses to stress in rodents (Belz et al. Citation2003) and should become a focus of future work. Therapeutic interventions might re-stimulate or inhibit those same neurons and genes epigenetically programmed earlier in life and reverse these changes. However, timing, quality and duration of such environmental stimulations might be highly specific and context dependent with regard to the exact nature of the early life stressor. Moreover, recent studies in the literature are challenging the static view of a locked state for DNA methylation, demonstrating that DNA methylation remains an active process in postmitotic cells (i.e. neurons). For example, DNMTs are expressed and regulated in the adult brain (Levenson et al. Citation2006; Brown et al. Citation2008; Zhang et al. Citation2010) and evidence continues to mount that epigenetic mechanisms are able to dynamically control changes in gene activity throughout the lifespan as a result of exposure to a variety of environmental factors (Miller and Sweatt Citation2007; Lubin et al. Citation2008; Westberry et al. Citation2008; Anier et al. Citation2010).
A further option could be the use of ‘epigenetic drugs’ targeting DNA methylation and HDAC enzymes, although those currently available drugs suffer from a lack of specificity at both the levels of tissue and the genome. Nevertheless, it is notable that the frequently used mood stabilizer valproate has been shown to modulate the epigenome by inhibiting HDACs (for review, see Machado-Vieira et al. Citation2010) and can promote demethylation in the brain (Tremolizzo et al. Citation2002; Dong et al. Citation2010; Perisic et al. Citation2010). This field may yet develop further with research involving the inhibition of specific histone-modifying enzymes, such as G9a, that may be selective enough to better target addiction or mental illness (Maze et al. Citation2010). Conceptually, DNA methylation and histone marks appear interdependent and treatments targeting histone acetylation might influence DNA methylation as well (Szyf Citation2009). In fact, this approach might seem more effective than currently available drugs in inhibiting DNA methylation (e.g. 5-azacytidine) which rely on incorporation into the DNA of mitotically active cells. Though such chromatin- and DNA methylation-targeting drugs are proving highly attractive to cancer therapeutics, it remains to be seen whether they have any real potential in the field of psychiatric diseases.
Another interesting approach to regulate methylation involves treatment with the methyl donor l-methionine, or folic acid supplementation during gestation. Although variable effects in gene expression and behaviour have been reported (Waterland and Jirtle Citation2003; Weaver et al. Citation2005; Burdge et al. Citation2009; LaPlant et al. Citation2010; Lillycrop et al. Citation2010), the mechanisms of how such treatment could target and induce such effects in specific regions of the brain are still yet not properly understood.
In sum, understanding how early life experiences can give rise to lasting epigenetic memories conferring increased risk for mental disorders, how they are maintained and how they could be reversed, is rapidly becoming a focus of modern psychiatry and should pave new guidelines for timely therapeutic interventions and the development of new pharmacological leads.
Declaration of interest: This work was funded by the European Union (CRESCENDO—European Union Contract number LSHM-CT-2005-018652) and the Deutsche Forschungsgemeinschaft (SP 386/4-2 to D.S.). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Aguilera G, Rabadan-Diehl C. 2000. Vasopressinergic regulation of the hypothalamic–pituitary–adrenal axis: Implications for stress adaptation. Regul Pept. 96:23–29.
- Albert PR. 2010. Epigenetics in mental illness: Hope or hype?. J Psychiatry Neurosci. 35:366–368.
- . 2007. EpigeneticsAllis C, Jenuwein T, Reinberg D, Caparros ML. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. 2010. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 35:2450–2461.
- Belz EE, Kennell JS, Czambel RK, Rubin RT, Rhodes ME. 2003. Environmental enrichment lowers stress-responsive hormones in singly housed male and female rats. Pharmacol Biochem Behav. 76:481–486.
- Bracaglia G, Conca B, Bergo A, Rusconi L, Zhou Z, Greenberg ME, Landsberger N, Soddu S, Kilstrup-Nielsen C. 2009. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 10:1327–1333.
- Brown SE, Weaver ICG, Meaney MJ, Szyf M. 2008. Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci Lett. 440:49–53.
- Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, Hanson MA. 2009. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr. 139:1054–1060.
- Chahrour M, Zoghbi HY. 2007. The story of Rett syndrome: From clinic to neurobiology. Neuron. 56:422–437.
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 302:885–889.
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 19:269–301.
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. 2010. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci. 13:1128–1136.
- Dong E, Chen Y, Gavin DP, Grayson DR, Guidotti A. 2010. Valproate induces DNA demethylation in nuclear extracts from adult mouse brain. Epigenetics. 5:730–735.
- Eccleston A, DeWitt N, Gunter C, Marte B, Nath D. 2007. Epigenetics, nature insight. Nature. 447:395–440.
- Engelmann M, Landgraf R, Wotjak CT. 2004. The hypothalamic-neurohypophysial system regulates the hypothalamic-pituitary-adrenal axis under stress: an old concept revisited. Front Neuroendocrinol. 25:132–149.
- Flavell SW, Greenberg ME. 2008. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 31:563–590.
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. 2008. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 359:61–73.
- Goekoop JG, de Winter R, Wolterbeek R, Wiegant V. Support for two increased vasopressinergic activities in depression at large and the differential effect of antidepressant treatment. J Psychopharmacol. 2010 doi: 10.1177/0269881110372549.
- Green JG, McLaughlin KA, Berglund PA, Gruber MJ, Sampson NA, Zaslavsky AM, Kessler RC. 2010. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: Associations with first onset of DSM-IV disorders. Arch Gen Psychiatry. 67:113–123.
- Greer PL, Greenberg ME. 2008. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron. 59:846–860.
- Heim C, Nemeroff CB. 2001. The role of childhood trauma in the neurobiology of mood and anxiety disorders: Preclinical and clinical studies. Biol Psychiatry. 49:1023–1039.
- Heim C, Nemeroff CB. 2002. Neurobiology of early life stress: Clinical studies. Semin Clin Neuropsychiatry. 7:147–159.
- Heim C, Newport DJ, Wagner D, Wilcox MM, Miller AH, Nemeroff CB. 2002. The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: A multiple regression analysis. Depress Anxiety. 15:117–125.
- Heim C, Plotsky PM, Nemeroff CB. 2004. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology. 29:641–648.
- Heim C, Mletzko T, Purselle D, Musselman DL, Nemeroff CB. 2008. The dexamethasone/corticotropin-releasing factor test in men with major depression: Role of childhood trauma. Biol Psychiatry. 63:398–405.
- Hochberg Z, Feil R, Constancia M, Fraga M, Junien C, Carel JC, Boileau P, Le Bouc Y, Deal CL, Lillycrop K, Scharfmann R, Sheppard A, Skinner M, Szyf M, Waterland RA, Waxman DJ, Whitelaw E, Ong K, Albertsson-Wikland K. 2011. Child health, developmental plasticity, and epigenetic programming. Endocr Rev. 32:159–224.
- Katz LC, Shatz CJ. 1996. Synaptic activity and the construction of cortical circuits. Science. 274:1133–1138.
- Klose RJ, Bird AP. 2006. Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci. 31:89–97.
- Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. 2005. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 19:667–678.
- Landgraf R. 2006. The involvement of the vasopressin system in stress-related disorders. CNS Neurol Disord Drug Targets. 5:167–179.
- LaPlant Q, Vialou V, Covington HEIII, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ. 2010. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 13:1137–1143.
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. 2006. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 281:15763–15773.
- Lillycrop KA, Rodford J, Garratt ES, Slater-Jefferies JL, Godfrey KM, Gluckman PD, Hanson MA, Burdge GC. 2010. Maternal protein restriction with or without folic acid supplementation during pregnancy alters the hepatic transcriptome in adult male rats. Br J Nutr. 103:1711–1719.
- Lubin FD, Roth TL, Sweatt JD. 2008. Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. J Neurosci. 28:10576–10586.
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. 2009. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 10:434–445.
- Machado-Vieira R, Ibrahim L, Zarate CAJr. Histone deacetylases and mood disorders: Epigenetic programming in gene–environment interactions. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00203.x.
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. 2003. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 302:890–893.
- Maze I, Covington HE3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. 2010. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 327:253–257.
- McCormick CM, Mathews IZ, Thomas C, Waters P. 2010. Investigations of HPA function and the enduring consequences of stressors in adolescence in animal models. Brain Cogn. 72:73–85.
- McGowan PO, Sasaki A, Huang TCT, Unterberger A, Suderman M, Ernst C, Meaney MJ, Turecki G, Szyf M. 2008. Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS ONE. 3:e2085.
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. 2009. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 12:342–348.
- Meaney MJ, Ferguson-Smith AC. 2010. Epigenetic regulation of the neural transcriptome: The meaning of the marks. Nat Neurosci. 13:1313–1318.
- Miller CA, Sweatt JD. 2007. Covalent modification of DNA regulates memory formation. Neuron. 53:857–869.
- Murgatroyd C, Spengler D. unpublished 2011.
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. 2009. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 12:1559–1566.
- Murgatroyd C, Wu Y, Bockmühl Y, Spengler D. Genes learn from stress: How infantile trauma programs us for depression. Epigenetics. 2010a; 5:194–199.
- Murgatroyd C, Wu Y, Bockmühl Y, Spengler D. The Janus face of DNA methylation in aging. Aging-US. 2010b; 2:107–110.
- Murgatroyd C, Hoffman A, Spengler D. 2011. In vivo ChIP of brain tissues. Methods Mol Biol 809: 2011.
- Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M, Holsboer F, Rein T, Zschocke J. 2010. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology. 35:792–805.
- Pryce CR, Feldon J. 2003. Long-term neurobehavioural impact of the postnatal environment in rats: Manipulations, effects and mediating mechanisms. Neurosci Biobehav Rev. 27:57–71.
- Satre JP. 1964. Les Mots. Paris: Gallimard254–255.
- Szyf M. 2009. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 49:243–263.
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, Sun YE. 2009. Phosphorylation of MeCP2 at serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci USA. 106:4882–4887.
- Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A. 2002. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci USA. 99:17095–17100.
- Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y. 2011. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 69:359–372.
- Veenema AH, Blume A, Niederle D, Buwalda B, Neumann ID. 2006. Effects of early life stress on adult male aggression and hypothalamic vasopressin and serotonin. Eur J Neurosci. 24:1711–1720.
- Waterland RA, Jirtle RL. 2003. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 23:5293–5300.
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004. Epigenetic programming by maternal behaviour. Nat Neurosci. 7:847–854.
- Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, Szyf M. 2005. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: Altering epigenetic marking later in life. J Neurosci. 25:11045–11054.
- Westberry JM, Prewitt AK, Wilson ME. 2008. Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience. 152:982–989.
- Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. 2010. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 30:13130–13137.
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 52:255–269.