Abstract
Activation of the in vivo stress response can facilitate antibacterial host defenses. One possible mechanism for this effect is stress-induced release of heat shock protein 72 (Hsp72) into the extracellular environment. Hsp72 is a ubiquitous cellular protein that is up-regulated in response to cellular stress, and modulates various aspects of immune function including macrophage inflammatory/bactericidal responses and T-cell function when found in the extracellular environment. The current study tested the hypothesis that in vivo extracellular Hsp72 (eHsp72) at the site of inflammation contributes to stress-induced restricted development of bacteria, and facilitated recovery from bacteria-induced inflammation, and that this effect is independent of alpha beta (αβ) T cells. Male F344 rats were exposed to either inescapable electrical tail-shocks or no stress, and subcutaneously injected with Escherichia coli (ATCC 15746). The role of eHsp72 was investigated by Hsp72-immunoneutralization at the inflammatory site. The potential contribution of T cells was examined by testing male athymic (rnu/rnu) nude rats lacking mature αβ T cells and heterozygous thymic intact control (rnu/+) rats. The results were that stressor exposure increased plasma concentrations of eHsp72 and facilitated recovery from bacterial inflammation. Immunoneutralization of eHsp72 at the inflammatory site attenuated this effect. Stressor exposure impacted bacterial inflammation and eHsp72 equally in both athymic and intact control rats. These results support the hypothesis that eHsp72 at the site of inflammation, and not αβ T cells, contributes to the effect of stressor exposure on subcutaneous bacterial inflammation.
Introduction
Acute activation of the stress response modulates many aspects of immunity (Ader Citation2007). The nature and consequence (i.e. adaptive vs. maladaptive) of the immunomodulatory impact of stressor exposure depends on a variety of factors including subject variables (age, gender, personality, and fitness status), stressor features (intensity, controllability, and duration), and the immunological measure. Hence, the immunological impact of a single well-characterized laboratory stressor (100, 1.6 mÅ, 5 s, av. 60-s random inter-trial interval tail-shocks) in a specific age, gender, and species of subject (adult male rats) includes elevated plasma and tissue inflammatory cytokines (Nguyen et al. Citation2000; Johnson et al. Citation2005), decreased mitogenic and antigenic stimulated T-cell cytokine production [interferon-gamma (IFNγ) and interleukin-2 (IL2)] and proliferation (Fleshner et al. Citation1995), increased acute-phase protein levels (Deak et al. Citation1997), and reduced in vivo antibody responses to neo-protein antigens (Colagiovanni et al. Citation2000; Fleshner et al. Citation2001; Moraska and Fleshner Citation2001; Kennedy et al. Citation2005).
Tail-shock exposure also facilitates the killing/clearing of a subcutaneous bacterial challenge that results in reduced bacteria-induced inflammation (Deak et al. Citation1999; Campisi et al. Citation2002; Campisi and Fleshner Citation2003), an effect that may be indicative of an adaptive consequence of stressor exposure. Specifically, rats exposed to tail-shock stress immediately prior to subcutaneous injections of Escherichia coli have reduced E. coli loads measured 2, 4, and 6 h after the in vivo bacteria challenge, and restricted inflammation diameters compared with control rats not exposed to stress (Campisi et al. Citation2003b). In addition, tail-shock stressed rats challenged with E. coli require fewer days to resolve the inflammation and suffer less body weight loss due to the E. coli challenge (Campisi et al. Citation2003b). The impact of tail-shock stress on the host response to E. coli likely involves facilitated antimicrobial cytokines and nitric oxide (NO) production at the site of inflammation (Campisi et al. Citation2002, Citation2003b). This is based on the observations that stressed rats challenged with bacteria have higher levels of NO and IL-1β at the inflammatory site and in vivo blockade of NO reduces the effect of tail-shock stress on facilitated inflammation resolution (Campisi et al. Citation2002). In addition, although tail-shock stress results in an increase in adrenal hormone secretion and sympathetic activation, it is unlikely that the stress-induced reduced bacteria-induced inflammation is a result of elevated corticosterone levels because adrenalectomy does not reduce this effect (Deak et al. Citation1999; Campisi et al. Citation2002; Campisi and Fleshner Citation2003). The current study expanded these observations to further investigate what additional factor(s) contribute to the impact of stress on bacterial inflammation. Specifically, the possible role of heat shock proteins (Hsps) and/or alpha beta (αβ) T cells was investigated.
Hsps are ubiquitous cellular proteins found in nearly all eukaryotic cells and are categorized into several families based on molecular weight (Morimoto Citation1994; Kregel Citation2002). When these proteins are intracellular, they primarily function as cellular chaperones and assist in the repair and transport of damaged proteins (Kiang and Tsokos Citation1998). More recently, however, it has become clear that extracellular Hsps can modulate immunity (Kaufmann Citation1990; Pockley and Multhoff Citation2008). Although there is evidence for immunomodulatory functions of several Hsps, our focus is on extracellular Hsp72 (eHsp72).
Hsp72 is a member of a family of closely related proteins termed the Hsp70 family. The Hsp70 family includes the constitutive 73-kDa protein (Hsc73) and a highly stress-inducible 72-kDa protein (Hsp72; Morimoto Citation1994; Hartl Citation1996). Intracellular Hsp72 level has been reported to increase after exposure to a variety of cellular and whole organism stressors including heat (Kregel Citation2002), tail-shock (Campisi et al. Citation2003c; Nickerson et al. Citation2006), and restraint (Udelsman et al. Citation1991; Udelsman et al. Citation1993). In addition, there is evidence that a variety of stressors can stimulate Hsp72 expression on the cell surface (Fehrenbach et al. Citation2000; Niess et al. Citation2002) and/or release into the extracellular environment. In humans, for example, eHsp72 level in the blood is increased after intense exercise (Febbraio and Koukoulas Citation2000; Walsh et al. Citation2001; Febbraio et al. Citation2002; Whitham and Fortes Citation2008), psychological work stress (Bugajska et al. Citation2008), and electrical hand shocks (Fleshner et al. Citation2006b). Interestingly, Pittet et al. (Citation2002) reported that the increase in eHsp72 found in the blood of trauma patients correlated with survival, such that the higher the concentration of eHsp72 the better the chances of survival. In rats, eHsp72 is also increased in the blood after electrical tail-shocks (Campisi et al. Citation2003b; Fleshner et al. Citation2003, Citation2004, Citation2006a), psychological predatory stress (Fleshner et al. Citation2004), and intense exercise (Brown et al. Citation2007). eHsp72 released into the blood after tail-shock is dependent on alpha-1 adrenergic receptor stimulation because administration of an alpha-1 adrenergic receptor antagonist blocked the effect (Johnson et al. Citation2005; Johnson and Fleshner Citation2006). Importantly, eHsp72 is also found in high concentration at the site of bacterial inflammation in tail-shock exposed, but not non-stressed controls (Fleshner et al. Citation2006b).
Endogenous extracellular or cell surface expressed Hsp72 can stimulate many aspects of immunity (Kaufmann Citation1990; Multhoff and Botzler Citation1998; Yoo et al. Citation2000; Prohaszka et al. Citation2002; Srivastava Citation2002; Campisi et al. Citation2003b; van Eden et al. Citation2003; Prohaszka and Fust Citation2004; Quintana and Cohen Citation2005; Johnson and Fleshner Citation2006; Calderwood et al. Citation2008; Pockley and Multhoff Citation2008). For example, eHsp72 stimulates inflammatory cytokine production from macrophages (Asea Citation2003; Campisi et al. Citation2003b), facilitates natural killer (NK) cell recognition and killing of cancer cells (Multhoff et al. Citation1995; Multhoff Citation1997, Citation2002), stimulates antigen presentation to T cells (Srivastava et al. Citation1994; Wells et al. Citation1998; Lehner et al. Citation2000; Noessner et al. Citation2002; Hickman-Miller and Hildebrand Citation2004; Lehner et al. Citation2004), and may bind in vivo to endogenous lipopolysaccharide (LPS) and augment immune responses by facilitating transfer of LPS to the toll-like receptor (TLR) 4-MD2 leading to improved signal transduction and cytokine responses (Quintana and Cohen Citation2005; Fleshner et al. Citation2007).
Thus, given the previous observations that tail-shock increases Hsp72 level in the blood and at the site of bacterial inflammation; and Hsp72 restricts the development of E. coli-induced inflammation when injected into the bacterial inflammatory site of a non-stressed control (Campisi et al. Citation2003b) and that the effect may depend on NO (Campisi et al. Citation2002), the present study tested the hypothesis that eHsp72 at the site of inflammation contributes to the effect of tail-shock stress on bacterial inflammation. eHsp72 was neutralized by a subcutaneous injection of the anti-Hsp70-Ab46 antibody into the inflammatory site. The anti-Hsp70-Ab46 antibody blocks several eHsp72 immune responses, including the stimulatory effect of Hsp72 on macrophage NO production (Svensson et al. Citation2006). Thus, we expected a reduction of the impact of stress on bacterial inflammation after eHsp72 immunoneutralization and not after control antibody. This effect should only occur in previously stressed rats, since only stressed rats have increased eHsp72 in the blood and at the bacterial inflammatory site (Fleshner et al. Citation2006b).
A second experiment examined the possible contribution of T cells to this effect. T cells are critical players in many immune responses including skin antimicrobial responses (Kolls et al. Citation2008), generation of fever (Wang et al. Citation2010), and types of skin inflammation [e.g. delayed-type hypersensitivity (DTH; Dhabhar and McEwen Citation1996)], and can be modulated by Hsp72. For example, a recent study demonstrated that stress mobilizes Hsp72 to the cell surface of dendritic cells (DC) modulating DC–T cell interaction and homeostasis of memory T cells involved in mediating fever (Wang et al. Citation2010). The stress-induced proliferation of memory T cells is of physiological significance given that fever is a common response of the body to infection (Wang et al. Citation2010). Additionally, it has been reported that endogenous eHsp72 may induce a population of anti-inflammatory T regulatory cells (Treg; van Eden et al. Citation2005). It is possible, therefore, that the reduction in bacterial inflammation produced by tail-shock is due to the proliferation of memory T cells which, in turn, modulate fever, or perhaps is due to the eHsp72 stimulation of anti-inflammatory Treg cells. To begin to test these hypotheses, we examined the effect of stress in the nude rat.
The homozygous athymic nude rat (rnu/rnu) lacks a normal thymus and mature and functioning αβ T cells. In contrast, the heterozyote rat (rnu/+) has a normal thymus and functioning αβ T cells and serves as the control. Athymic nude rats are ideal to use for this study because in spite of a lack of mature αβ T cells, they have normal numbers of, and properly functioning granulocytes, macrophages, NK cells, erythrocytes, B cells, and basal antibody (Rolstad Citation2001). If stress-induced restriction of bacterial inflammation is mediated by or dependent on αβ T cells, then nude rats should fail to demonstrate the effect.
Methods
Animals
Adult male viral-free Fischer 344 (F344), athymic (rnu/rnu) nude, and control heterozygote intact (rnu/+) rats (200–300 g; Harlan Labs, Indianapolis, IN, USA) were individually caged in Plexiglas cages (60 cm × 30 cm × 24 cm) with food and water available ad libitum. The rat colonies were maintained in a pathogen-free barrier facility with a 12:12 h light:dark cycle (lights on 06:00–18:00 h). Rats were given at least 1 week to habituate to the colonies prior to experimentation. Body weights were monitored daily. All rats were handled and weighed each day for at least 2 days before each study began. Care and use of the rats were in accordance with protocols approved by the University of Colorado Institutional Animal Care and Use Committee.
Inescapable tail-shock stress procedure
Rats either remained undisturbed in their home cages as no stress controls or were exposed to inescapable tail-shock as described previously (Fleshner et al. Citation1998; Campisi et al. Citation2002). Rats in the stress condition were transported to an adjacent room and placed in Plexiglas restraining tubes (15 cm × 7 cm). Contact beams were placed on the tail of the rat, and 100 1.6-mÅ tail-shocks (5-s duration, variable inter-trial interval ∼60 s; range 30–90 s) were administered. The total tail-shock session lasted approximately 100 min. All rats were shocked between 08:00 and 10:00 h. Importantly, this acute laboratory stressor has been previously reported to reduce bacterial load and to facilitate recovery following bacterial challenge (Deak et al. Citation1999; Campisi et al. Citation2002, Citation2003b; Fleshner et al. Citation2002; Campisi and Fleshner Citation2003).
Bacterial culture
E. coli (ATCC 15746) were purchased from American Type Culture Collection (Bethesda, MD, USA), re-hydrated, and grown overnight to maximal densities in 30 ml of brain–heart infusion (BHI; DIFCO Laboratories, Detroit, MI, USA) at 37°C, 95% air+5% CO2. Cultures were then aliquoted into 1.0 ml of BHI supplemented with 10% glycerol and frozen in sterile vials at − 70°C. These vials constituted the stock cultures. All experiments utilized bacteria from these stock cultures. One day prior to experimentation, stock cultures were thawed and cultured (37°C, 95% air+5% CO2) overnight (∼19 h) in 35 ml of BHI. The number of bacteria in cultures was quantified by extrapolating from previously determined growth curves (Deak et al. Citation1999; Campisi et al. Citation2002). Cultures were then centrifuged for 15 min at 1200g, supernatants discarded, and bacteria were re-suspended in sterile phosphate-buffered saline. All rats were injected subcutaneously with 2.5 × 108 colony-forming units (CFU) of E. coli because previous work in our laboratory determined that this dose produces a reliable inflammatory response (Deak et al. Citation1999). Although gram-positive bacteria such as Staphylococcus aureus are more likely to infect the skin than gram-negative bacteria such as E. coli, previous work (Deak et al. Citation1999) indicated that a subcutaneous injection of E. coli produces a more consistent and robust inflammatory response than did S. aureus. Furthermore, administration of gram-positive bacteria to the skin of a rodent introduces issues of endogenous contamination because opportunistic gram-positive bacteria infect the area making quantification of the administered bacteria difficult (Rojas et al. Citation2002).
Bacterial challenge and in vivo inflammation assessment
Inflammation diameter assessment has been described previously (Deak et al. Citation1999; Campisi et al. Citation2002, Citation2003b; Fleshner et al. Citation2002; Campisi and Fleshner Citation2003). Briefly, one day prior to injection, an area on the rat's dorsal surface measuring approximately 5.0 cm × 7.0 cm was shaved so that inflammation could easily be quantified in the F344 rats (shaving was not necessary for nude rats). On the day of experimentation, injections were given subcutaneously in a volume of 250 μl at the center of the back immediately following tail-shocks or no stress control treatment. The diameter of inflammation (to 0.1 mm) was measured hourly and/or daily after challenge until resolution. Measurements were taken between 10:00 and 11:00 h using a digital caliper. All measurements were made on the same region of the dorsum by an independent observer who was blind to the experimental condition of the rat.
In vivo immunoneutralization of Hsp72
Male F344 rats (6/group) were either exposed to tail-shock stress or remained in their home cages. Immediately after stressor termination, rats were injected subcutaneously with either E. coli + anti-Hsp70-Ab46 (84.0 μg/rat provided by Dr. Alexzander Asea) or E. coli + isotype control (Sigma, St. Louis, MO, USA) isotype control mouse immunoglobulin G1 (IgG1; 84.0 μg/rat, control Ab). To maintain a high concentration of anti-Hsp70-Ab46 at the site of inflammation, antibodies plus E. coli were suspended in incomplete Freund's vehicle as described previously (Campisi et al. Citation2003b). Inflammation diameters were measured as described above.
Effects of T-cell depletion on stress-induced facilitation of bacterial inflammation
Athymic nude (rnu/rnu) and control (rnu/+) rats were randomly assigned to tail-shock (n = 8/strain of animal) or home cage no stress control (n = 8/strain of rat) groups. Immediately following tail-shock or control treatment, all rats were injected with 2.5 × 108 CFU of E. coli and inflammation was measured as above.
Extracellular Hsp72
In a subset of F344, athymic nude (rnu/rnu) and control (rnu/+) rats (n = 6 per group), plasma concentrations of eHsp72 were measured using enzyme-linked immunosorbent assay (ELISA, Assay Design, Ann Arbor, MI, USA and StressGen Biotechnologies, Victoria, BC, Canada; ). Minimum detection of eHsp72 was 0.20 ng/ml with an inter-assay precision of 12.8% and an intra-assay precision of 3.9%. Trunk blood samples from rats exposed to tail-shock or no stress were collected immediately after stressor termination via decapitation. Plasma was retrieved from the ethylenediaminetetraacetic acid-treated blood after centrifugation and rapidly frozen for future ELISA analyses.
Data analysis
Data from studies involving multiple time measures were analyzed by repeated measures analysis of variance (RMANOVA), while all other data were analyzed by ANOVA to determine if main effects were present. If significant time × main effect interactions (RMANOVA) or main effects (ANOVA) were present, then Fisher's protected least significant difference (PLSD) post-hoc analyses were conducted. Alpha was set at 0.05. All figures are shown with group means and standard error (SE) of mean bars.
Results
In vivo immunoneutralization of eHsp72
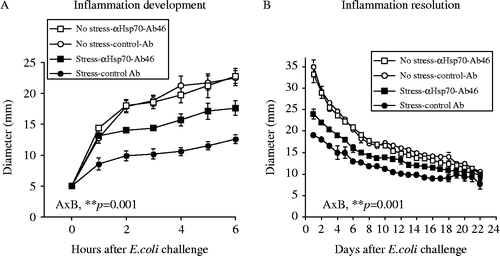
Subcutaneous bacterial challenge produced a time-dependent increase in inflammation (). Similar to our previous reports, exposure to tail-shock prior to bacterial challenge constrained the development () and facilitated the resolution () of the inflammation. Immunoneutralization of Hsp72 attenuated the effect of stress. Control antibody injection had no effect. RMANOVA revealed main effects of tail-shock and antibody on both the development , F(1,18) = 60.7, p ≤ 0.0001] and the resolution , F(1,20) = 120.0, p ≤ 0.0001] of the bacterial inflammation. Also supported by RMANOVA was a main effect of antibody on both the development , F(1,18) = 8.8, p ≤ 0.001] and the resolution , F(1,20) = 5.0, p ≤ 0.05] of inflammation. Importantly, there was also a reliable stress × antibody interaction on the development , F(1,18) = 9.1, p ≤ 0.001] and the resolution , F(1,20) = 11.5, p = 0.001] of bacterial inflammation, indicating that anti-Hsp70-Ab46 antibody at the site of inflammation attenuated the effect of stress.
Figure 1. Adult, male F344 rats were either exposed to 100, 5-s, 1.6 mÅ, inescapable tail-shocks (stress) or remained in their home cages as no stress controls. Immediately following stressor termination, all rats were injected subcutaneously with ∼2.5 × 08 CFU of E. coli and either anti-Hsp70-Ab46 or mouse IgG1 control antibody. Inflammation diameter was measured hourly (A) to assess development (0–12 h) and daily (B) to assess resolution (1–25 days) after E. coli challenge. RMANOVA revealed a main effect of tail-shock and anti-Hsp70-Ab46 on both inflammation development (A) and resolution (B; p's < 0.0001). There was a main effect of antibody on inflammation development (A; p ≤ 0.001) and resolution (B; p ≤ 0.05). A stress × antibody interaction (A × B) was found for development (A; p < 0.001) and resolution (B; p < 0.001) of inflammation. Data are group mean ± SE of the mean; 4–8 rats/group. CFU, colony-forming units; Hsp, heat shock protein; Ig, immunoglobulin.
Effect of stress on plasma concentrations of eHsp72
In adult male F344 rats, tail-shock stress increased plasma eHsp72 concentration [to mean ± standard error of the mean (SEM): 1848 ± 302 pg/ml vs. control mean ± SEM: 87 ± 7 pg/ml; F(1,8) = 42.85, p = 0.0001], replicating our previous observations (Campisi et al. Citation2003b; Fleshner et al. Citation2003, Citation2004, Citation2006b).
Effects of stress on facilitation of bacterial inflammation in athymic rats
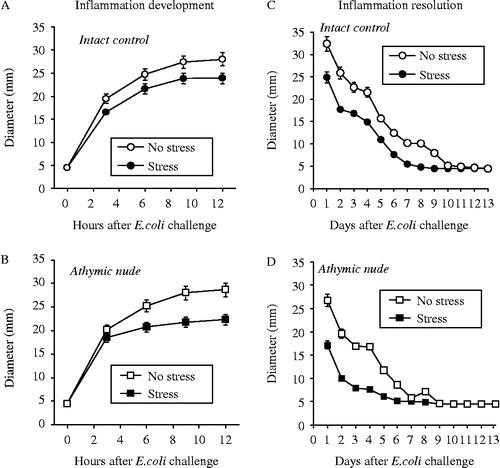
The inflammatory response of the athymic stressed rats was restricted compared to the non-stressed controls. Both Rnu/+ (intact control) and athymic nude (rnu/rnu) rats injected with E. coli immediately following tail-shocks had significant restrictions in the hourly development of inflammation over the course of the initial 12 h following injection (). RMANOVA revealed a main effect of tail-shocks on the development of inflammation in Rnu/+ [intact control; , F(4,56) = 195.3, p ≤ 0.001] and athymic nude (rnu/rnu) rats , F(4,56) = 187.9, p ≤ 0.001]. When inflammation diameter was examined 24 h following injection (i.e. day 1 following E. coli challenge), and daily thereafter until resolution occurred (), significant differences in resolution were observed between rats exposed to stress and controls. Rnu/+ (intact control) rats that were injected with E. coli immediately following tail-shock had a 23% reduction [F(1,14) = 222, p = 0.0067] in inflammation based on diameter measures on day 1 () and athymic nude (rnu/rnu) rats injected with E. coli immediately following tail-shock had a 35% reduction [F(1,14) = 370.5, p = 0.0008] in inflammation based on diameter measures on day 1 (). Both Rnu/+ (intact control) rats and athymic nude (rnu/rnu) rats that were injected with E. coli immediately following tail-shock showed faster resolution of inflammation than non-stressed controls (). Indeed, exposure to stress in the athymic nude rats resulted in the greatest restriction and fastest recovery compared to all other groups, as supported by a group × strain × time interaction [F(13,364) = 2.064, p = 0.01].
Figure 2. Adult, male rnu+ (A, C) and rnu/rnu (B, D) athymic nude rats were either exposed to 100, 5 s, 1.6 mÅ, inescapable tail-shocks (stress) or remained in their home cages as no stress controls. Immediately following stressor termination, all rats were injected subcutaneously with ∼2.5 × 108 (CFU) of E. coli. Inflammation diameter was measured hourly to assess development (0–12 h) and daily to assess resolution (1–25 days) after E. coli challenge. Rnu/+ (A) and athymic nude (rnu/rnu) (B) rats injected with E. coli immediately following tail-shock stress showed significantly reduced hourly development of inflammation over the course of the initial 12 h following injection (A,B). RMANOVA revealed a main effect of tail-shocks on the development of inflammation in Rnu/+ (A, p ≤ 0.001) and rnu/rnu rats (B, p ≤ 0.001). RMANOVA revealed that Rnu/+ rats that were injected with E. coli immediately following tail-shocks had a significant reduction in inflammation diameter measured 24 h following injection (i.e. on day 1; p = 0.0067; Figure 2C), and athymic nude (rnu/rnu) rats injected with E. coli immediately following stress also had a significant reduction (p = 0.0008; Figure 2D) in inflammation based on diameter measured on day 1. Exposure to stress in the athymic nude rats resulted in the greatest restriction and fastest recovery as compared to all other groups (group × strain × time interaction; p = 0.01). Data are group mean ± SE of the mean; 4–8 rats/group. CFU, colony-forming units.
Effect of stress on plasma concentrations of eHsp72 in athymic nude (rnu/rnu) and intact control (rnu/+) rats
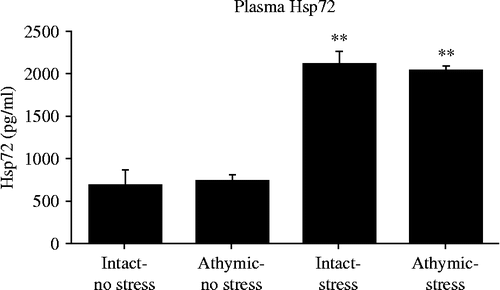
In both athymic nude rats and heterozygous intact control rats, tail-shock (stress) increased plasma eHsp72 concentration compared to no stress controls ; F(3,12) = 18.9, p = 0.0001], replicating our previous observations in F344 rats (Campisi et al. Citation2003c; Nickerson et al. Citation2006) and extending this observation to two additional strains of rats.
Figure 3. Adult, male rnu+ and rnu/rnu athymic nude rats were either exposed to 100, 5 s, 1.6 mÅ, inescapable tail-shocks (stress) or remained in their home cages as no stress controls. Immediately following stressor termination, all rats were rapidly decapitated and plasma collected. Hsp72 concentrations were measured using ELISA. ANOVA indicated that both athymic nude rats and heterozygous intact control rats exposed to tail-shocks (stress) increased plasma eHsp72 concentration compared to no stress controls (p = 0.0001). Data are group mean ± SE of the mean; 4–8 rats/group. ELISA, enzyme-linked immunosorbent assay; eHsp, extracellular heat shock protein.
Discussion
The current study tested the hypothesis that eHsp72 at the inflammatory site, and not αβ T cells, mediates the effect of acute stressor exposure on bacterial inflammation. The results from the current study support our hypothesis as immunoneutralization of eHsp72 blocked the effect of stress, and exposure to acute stress was equally efficacious in both homozygote (rnu/run) athymic nude rats and heterozygote intact control (rnu/+) rats.
Consistent with previous reports, acute stressor exposure resulted in a restriction in inflammation development and a facilitation of inflammation recovery (Deak et al. Citation1999; Campisi et al. Citation2002, Citation2003b; Fleshner et al. Citation2002; Campisi and Fleshner Citation2003). Importantly, injection of anti-Hsp70-Ab, but not isotype control antibody, into the inflammatory site attenuated this effect. These data () support our hypothesis that Hsp72 at the inflammatory site plays a role in the effect of stress. We have previously reported that subcutaneous injection of E. coli results in dose-dependent inflammatory responses, i.e. the larger the number of CFU injected the larger the inflammation development (Deak et al. Citation1999; Campisi et al. Citation2003a). In addition, rats exposed to tail-shock stress compared to no stress immediately prior to subcutaneous injections of E. coli have reduced E. coli loads, restricted inflammation development, and a facilitation of inflammation recovery (Deak et al. Citation1999; Campisi et al. Citation2002, Citation2003b; Fleshner et al. Citation2002; Campisi and Fleshner Citation2003). Thus, there is a relationship between inflammation diameter and the quantity of bacteria present at the inflammatory site. The results from the present series of experiments, as well as previous results (Campisi et al. Citation2003b; Campisi and Fleshner Citation2003) indicate that Hsp72 at the inflammatory site modulates the killing and/or clearance of bacteria at the inflammatory site, resulting in restricted inflammation development.
Interestingly, it has been proposed that Hsp72 released by a wide variety of stressors may function as a “messenger of stress” or “danger signal” to the immune system (Colaco Citation1998; Chen et al. Citation1999; Asea et al. Citation2000; Ohashi et al. Citation2000; Todryk et al. Citation2000; Breloer et al. Citation2001; Bethke et al. Citation2002; Habich et al. Citation2002; Vabulas et al. Citation2002). The notion that in addition to pathogen associated signals, endogenous danger/alarm signals such as Hsp72 that are derived from stressed or damaged self can activate immune cells was initially proposed by Matzinger (Citation1994). The idea was originally controversial (Matzinger Citation1994, Citation1998), but is rapidly gaining acceptance (Davies et al. Citation2006; Kohl Citation2006; Manjili et al. Citation2006; Shi et al. Citation2006; Zedler and Faist Citation2006; El Mezayen et al. Citation2007; Yokota and Fujii Citation2010; Zampell et al. Citation2011).
How extracellular Hsp72 functions to prime or facilitate the innate antimicrobial response remains a topic of investigation? It seems clear, however, that endogenous Hsp72 released into the blood is likely not “naked.” Indeed, it has been previously suggested that considering the complex-forming ability and chaperoning function of Hsp72, in vivo “endotoxin-free” Hsp is probably non-existent (Prohaszka and Fust Citation2004). This observation, and the finding that exposure to a variety of physical and psychological stressors stimulates commensal bacterial translocation from the gut (Deitch and Berg Citation1987; Horton Citation1994; Katafuchi et al. Citation2003; Nettelbladt et al. Citation2003; Nazli et al. Citation2004; Velin et al. Citation2004; Bailey et al. Citation2006), has led us to speculate that perhaps some or all of the endogenous Hsp72 released by acute stress into the blood may be associated with LPS released from endogenous gut bacterial flora. Perhaps it is this Hsp72/LPS complex that extravasates across the leaky vasculature or is transported from the blood into the inflamed tissues that facilitates host antimicrobial defenses. A recent study demonstrated that the exposure to stress resulted in significant changes in composition, diversity, and number of gut microorganisms as well as increased circulating levels of IL-6 and monocyte chemoattractant protein-1 (Bailey et al. Citation2011). However, mice treated with an antibiotic cocktail prior to stressor exposure failed to demonstrate the previously observed stress-induced elevations in circulating inflammatory proteins, indicating a causal relationship between the gut microbiota and stress-induced elevations in circulating cytokines. Future research will test the contribution of Hsp72 in these observations.
Interestingly, Quintana and Cohen (Citation2005) suggested that endogenous Hsps may perform diverse functions in two alternative modes of inflammation: sterile inflammation, which results from endogenous stimuli and is necessary for body maintenance, and septic inflammation, which gives protection against environmental pathogens. These authors offer the intriguing idea that Hsp72/LPS complexes may augment immune responses by facilitating the transfer of LPS to the TLR4-MD2 leading to improved signal transduction and inflammatory cytokine responses. In our model, we have evidence supporting both functions of Hsp72. The release of Hsp72 by psychological/physical stressors in the absence of an infectious agent could be an example of sterile inflammation, whereas the accumulation of Hsp72 at the site of bacterial inflammation and its role in facilitating host antimicrobial responses could be an example of septic inflammation. Quintana and Cohen (Citation2005) also describe regulatory mechanisms induced by Hsp72 that could possibly control anti-inflammatory responses. Thus, it is conceivable that the upregulation of Hsp72 facilitates bacterial clearance through the regulation of pro- and anti-inflammatory mechanisms. Additional work is required before firm conclusions can be made.
To examine the role of mature T cells in our model, we utilized the homozygous athymic nude rat (rnu/rnu), which lacks a normal thymus and functionally mature αβ T cells (Rolstad Citation2001). These rats are particularly useful when examining the contribution of specific components of the immune system because, despite the absence of mature T cells, most aspects of innate immunity are intact (Rolstad Citation2001). Both athymic nude rats and heterozygous thymic intact control rats exposed to an acute stressor demonstrated a restriction in the maximum inflammatory response (), indicating that mature T cells are not necessary for this effect.
In addition to lending support to our hypothesis, these results also demonstrate that the effect of acute stress exposure is generalizable across different strains of rats. Previous research has indicated that F344 (Campisi et al. Citation2002, Citation2003c; Campisi and Fleshner Citation2003) and Sprague-Dawley (Deak et al. Citation1999; Fleshner et al. Citation2002) rats exposed to tail-shock demonstrate a restriction in inflammation, and the results of the current study extend these observations to the rnu/rnu athymic nude and rnu/+ strains. These data indicate that the effect of stress on inflammation is a generalizable phenomenon that occurs in a variety of rat strains.
In conclusion, the results of the current study indicate that the observed stress-induced restriction in inflammation is mediated, at least in part, by extracellular Hsp72 at the site of bacterial inflammation, and is not dependent on αβ T cells. The presence of a stress-induced restriction in inflammation in the absence of many features of acquired immunity indicates that the initial or immediate effect of stress is indeed an innate immune phenomenon.
Acknowledgment
We thank Dr Ben Greenwood for helpful discussions regarding the writing of the manuscript.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ader R. 2007. PsychoneuroimmunologyElsevier Academic Press.
- Asea A. 2003. Chaperokine-induced signal transduction pathways. Exerc Immunol Rev. 9:25–33.
- Asea A, Kabingu E, Stevenson MA, Calderwood SK. 2000. HSP70 peptide-bearing and peptide-negative preparations act as chaperokines. Cell Stress Chaperones. 5:425–431.
- Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. 2011. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav Immun. 25:397–407.
- Bailey MT, Engler H, Sheridan JF. 2006. Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol. 171:29–37.
- Bethke K, Staib F, Distler M, Schmitt U, Jonuleit H, Enk AH, Galle PR, Heike M. 2002. Different efficiency of heat shock proteins (HSP) to activate human monocytes and dendritic cells: Superiority of HSP60. J Immunol. 169:6141–6148.
- Breloer M, Dorner B, More SH, Roderian T, Fleischer B, von Bonin A. 2001. Heat shock proteins as “danger signals”: Eukaryotic Hsp60 enhances and accelerates antigen-specific IFN-gamma production in T cells. Eur J Immunol. 31:2051–2059.
- Brown DA, Johnson MS, Armstrong CJ, Lynch JM, Caruso NM, Ehlers LB, Fleshner M, Spencer RL, Moore RL. 2007. Short-term treadmill running in the rat: What kind of stressor is it?. J Appl Physiol. 103:1979–1985.
- Bugajska J, Widerszal-Bazyl M, Radkiewicz P, Pasierski T, Szulczyk GA, Zabek J, Wojciechowska B, Jedryka-Goral A. 2008. Perceived work-related stress and early atherosclerotic changes in healthy employees. Int Arch Occup Environ Health. 81:1037–1043.
- Calderwood SK, Gong J, Theriault JR, Mambula SS, Gray PJJr. 2008. Cell stress proteins: Novel immunotherapeutics. Novartis Found Symp. 291:115–131 discussion 131–140.
- Campisi J, Fleshner M. 2003. The role of extracellular Hsp72 in acute stress-induced potentiation of innate immunity in physically active rats. J Appl Physiol. 94:43–52.
- Campisi J, Hansen MK, O'Connor KA, Biedenkapp JC, Watkins LR, Maier SF, Fleshner M. Circulating cytokines and endotoxin are not necessary for the activation of the sickness or corticosterone response produced by peripheral E. coli challenge. J Appl Physiol. 2003a; 95:1873–1882.
- Campisi J, Leem TH, Fleshner M. 2002. Acute stress decreases inflammation at the site of infection. A role for nitric oxide. Physiol Behav. 77:291–299.
- Campisi J, Leem TH, Fleshner M. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones. 2003b; 8:272–286.
- Campisi J, Leem TH, Greenwood BN, Hansen MK, Moraska A, Higgins K, Smith TP, Fleshner M. Habitual physical activity facilitates stress-induced HSP72 induction in brain, peripheral and immune tissues. Am J Physiol. 2003c; 284:520–530.
- Chen HW, Chou FP, Lue SI, Hsu HK, Yang RC. 1999. Evidence of multi-step regulation of HSP72 expression in experimental sepsis. Shock. 12:63–68.
- Colaco CA. 1998. Towards a unified theory of immunity: Dendritic cells, stress proteins and antigen capture. Cell Mol Biol. 44:883–890.
- Colagiovanni DB, Suniga MA, Frazier JL, Edwards CK3rd, Fleshner M, McCay JA, White KLJr, Shopp GM. 2000. TNF-alpha blockade by a dimeric TNF type I receptor molecule selectively inhibits adaptive immune responses. Immunopharmacol Immunotoxicol. 22:627–651.
- Davies EL, Bacelar MM, Marshall MJ, Johnson E, Wardle TD, Andrew SM, Williams JH. 2006. Heat shock proteins form part of a danger signal cascade in response to lipopolysaccharide and GroEL. Clin Exp Immunol. 145:183–189.
- Deak T, Meriwether JL, Fleshner M, Spencer RL, Abouhamze A, Moldawer LL, Grahn RE, Watkins LR, Maier SF. 1997. Evidence that brief stress may induce the acute phase response in rats. Am J Physiol. 273:R1998–R2004.
- Deak T, Nguyen KT, Fleshner M, Watkins LR, Maier SF. 1999. Acute stress may facilitate recovery from a subcutaneous bacterial challenge. Neuroimmunomodulation. 6:344–354.
- Deitch EA, Berg R. 1987. Bacterial translocation from the gut: A mechanism of infection. J Burn Care Rehabil. 8:475–482.
- Dhabhar FS, McEwen BS. 1996. Stress-induced enhancement of antigen-specific cell-mediated immunity. J Immunol. 156:2608–2615.
- El Mezayen R, El Gazzar M, Seeds MC, McCall CE, Dreskin SC, Nicolls MR. 2007. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 111:36–44.
- Febbraio MA, Koukoulas I. 2000. HSP72 gene expression progressively increases in human skeletal muscle during prolonged, exhaustive exercise. J Appl Physiol. 89:1055–1060.
- Febbraio MA, Ott P, Nielsen HB, Steensberg A, Keller C, Krustrup P, Secher NH, Pedersen BK. 2002. Exercise induces hepatosplanchnic release of heat shock protein 72 in humans. J Physiol. 544:957–962.
- Fehrenbach E, Passek F, Niess AM, Pohla H, Weinstock C, Dickhuth HH, Northoff H. 2000. HSP expression in human leukocytes is modulated by endurance exercise. Med Sci Sports Exerc. 32:592–600.
- Fleshner M, Campisi J, Amiri L, Diamond DM. 2004. Cat exposure induces both intra- and extra-cellular HSP72: The role of adrenal hormones. Psychoneuroendocrinology. 29:1142–1152.
- Fleshner M, Campisi J, Deak T, Greenwood BN, Kintzel JA, Leem TH, Smith TP, Sorensen B. 2002. Acute stressor exposure facilitates innate immunity more in physically active than in sedentary rats. Am J Physiol Regul Integr Comp Physiol. 282:R1680–R1686.
- Fleshner M, Campisi J, Johnson JD. 2003. Can exercise stress facilitate innate immunity? A functional role for stress-induced extracellular Hsp72. Exerc Immunol Rev. 9:6–24.
- Fleshner M, Deak T, Nguyen KT, Watkins LR, Maier SF. 2001. Endogenous glucocorticoids play a positive regulatory role in the anti-keyhole limpet hemocyanin in vivo antibody response. J Immunol. 166:3813–3819.
- Fleshner M, Hermann J, Lockwood LL, Watkins LR, Laudenslager ML, Maier SF. 1995. Stressed rats fail to expand the CD45RC+CD4+ (Th1-like) T cell subset in response to KLH: Possible involvement of IFN-gamma. Brain Behav Immun. 9:101–112.
- Fleshner M, Johnson JD, Friedman J. 2007. Extracellular Hsp72: A double-edged sword for host defenseNew York, NY: Springer.
- Fleshner M, Nguyen KT, Cotter CS, Watkins LR, Maier SF. 1998. Acute stressor exposure both suppresses acquired immunity and potentiates innate immunity. Am J Physiol. 275:R870–R878.
- Fleshner M, Sharkey C, Nickerson M, Johnson ADEndogenous extracellular Hsp72 release is an adaptive feature of the acute stress response. In: Ader R, Felton DL, Cohen N. editors. Psychoneuroimmunology. San Diego, CA: Academic Press2006a.
- Fleshner M, Sharkey CM, Nickerson M, Johnson JDEndogenous extracellular Hsp72 release is an adaptive feature of the acute stress response. In: Ader R, Felton DL, Cohen N. editors. Psychoneuroimmunology. San Diego, CA: Academic Press2006b.
- Habich C, Baumgart K, Kolb H, Burkart V. 2002. The receptor for heat shock protein 60 on macrophages is saturable, specific, and distinct from receptors for other heat shock proteins. J Immunol. 168:569–576.
- Hartl FU. 1996. Molecular chaperones in cellular protein folding. Nature. 381:571–579.
- Hickman-Miller HD, Hildebrand WH. 2004. The immune response under stress: the role of HSP-derived peptides. Trends Immunol. 25:427–433.
- Horton JW. 1994. Bacterial translocation after burn injury: The contribution of ischemia and permeability changes. Shock. 1:286–290.
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Fleshner M. 2005. Adrenergic receptors mediate stress-induced elevations in extracellular Hsp72. J Appl Physiol. 99:1789–1795.
- Johnson JD, Fleshner M. 2006. Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J Leukoc Biol. 79:425–434.
- Katafuchi T, Takaki A, Take S, Kondo T, Yoshimura M. 2003. Endotoxin inhibitor blocks heat exposure-induced expression of brain cytokine mRNA in aged rats. Brain Res Mol Brain Res. 118:24–32.
- Kaufmann SH. 1990. Heat shock proteins and the immune response. Immunol Today. 11:129–136.
- Kennedy SL, Nickerson M, Campisi J, Johnson JD, Smith TP, Sharkey C, Fleshner M. 2005. Splenic norepinephrine depletion following acute stress suppresses in vivo antibody response. J Neuroimmunol. 165:150–160.
- Kiang JG, Tsokos GC. 1998. Heat shock protein 70 kDa: Molecular biology, biochemistry, and physiology. Pharmacol Ther. 80:183–201.
- Kohl J. 2006. Self, non-self, and danger: A complementary view. Adv Exp Med Biol. 586:71–94.
- Kolls JK, McCray PBJr, Chan YR. 2008. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 8:829–835.
- Kregel KC. 2002. Invited Review: Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol. 92:2177–2186.
- Lehner T, Bergmeier LA, Wang Y, Tao L, Sing M, Spallek R, van der Zee R. 2000. Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. Eur J Immunol. 30:594–603.
- Lehner T, Wang Y, Whittall T, McGowan E, Kelly CG, Singh M. 2004. Functional domains of HSP70 stimulate generation of cytokines and chemokines, maturation of dendritic cells and adjuvanticity. Biochem Soc Trans. 32:629–632.
- Manjili MH, Park JE, Facciponte JG, Wang XY, Subjeck JR. 2006. Immunoadjuvant chaperone, GRP170, induces ‘danger signals’ upon interaction with dendritic cells. Immunol Cell Biol. 84:203–208.
- Matzinger P. 1994. Tolerance, danger, and the extended family. Annu Rev Immunol. 12:991–1045.
- Matzinger P. 1998. An innate sense of danger. Semin Immunol. 10:399–415.
- Moraska A, Fleshner M. 2001. Voluntary physical activity prevents stress-induced behavioral depression and anti-KLH antibody suppression. Am J Physiol Regul Integr Comp Physiol. 281:R484–R489.
- Morimoto RI. 1994. The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Multhoff G. 1997. Heat shock protein 72 (HSP72), a hyperthermia-inducible immunogenic determinant on leukemic K562 and Ewing's sarcoma cells. Int J Hyperthermia. 13:39–48.
- Multhoff G. 2002. Activation of natural killer cells by heat shock protein 70. Int J Hyperthermia. 18:576–585.
- Multhoff G, Botzler C. 1998. Heat-shock proteins and the immune response. Ann NY Acad Sci. 851:86–93.
- Multhoff G, Botzler C, Wiesnet M, Muller E, Meier T, Wilmanns W, Issels RD. 1995. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer. 61:272–279.
- Nazli A, Yang PC, Jury J, Howe K, Watson JL, Soderholm JD, Sherman PM, Perdue MH, McKay DM. 2004. Epithelia under metabolic stress perceive commensal bacteria as a threat. Am J Pathol. 164:947–957.
- Nettelbladt CG, Katouli M, Bark T, Svenberg T, Mollby R, Ljungqvist O. 2003. Orally inoculated Escherichia coli strains colonize the gut and increase bacterial translocation after stress in rats. Shock. 20:251–256.
- Nguyen KT, Deak T, Will MJ, Hansen MK, Hunsaker BN, Fleshner M, Watkins LR, Maier SF. 2000. Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1beta protein response to acute stress. Brain Res. 859:193–201.
- Nickerson M, Kennedy SL, Johnson JD, Fleshner M. 2006. Sexual dimorphism of the intracellular heat shock protein 72 response. J Appl Physiol. 101:566–575.
- Niess AM, Fehrenbach E, Schlotz E, Sommer M, Angres C, Tschositsch K, Battenfeld N, Golly IC, Biesalski HK, Northoff H, Dickhuth HH. 2002. Effects of RRR-alpha-tocopherol on leukocyte expression of HSP72 in response to exhaustive treadmill exercise. Int J Sports Med. 23:445–452.
- Noessner E, Gastpar R, Milani V, Brandl A, Hutzler PJ, Kuppner MC, Roos M, Kremmer E, Asea A, Calderwood SK, Issels RD. 2002. Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol. 169:5424–5432.
- Ohashi K, Burkart V, Flohe S, Kolb H. 2000. Cutting edge: Heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 164:558–561.
- Pittet JF, Lee H, Morabito D, Howard MB, Welch WJ, Mackersie RC. 2002. Serum levels of Hsp 72 measured early after trauma correlate with survival. J Trauma. 52:611–617 discussion 617.
- Pockley AG, Multhoff G. 2008. Cell stress proteins in extracellular fluids: Friend or foe?. Novartis Found Symp. 291:86–95 discussion 96–100, 137–140.
- Prohaszka Z, Fust G. 2004. Immunological aspects of heat-shock proteins – the optimum stress of life. Mol Immunol. 41:29–44.
- Prohaszka Z, Singh M, Nagy K, Kiss E, Lakos G, Duba J, Fust G. 2002. Heat shock protein 70 is a potent activator of the human complement system. Cell Stress Chaperones. 7:17–22.
- Quintana FJ, Cohen IR. 2005. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol. 175:2777–2782.
- Rojas IG, Padgett DA, Sheridan JF, Marucha PT. 2002. Stress-induced susceptibility to bacterial infection during cutaneous wound healing. Brain Behav Immun. 16:74–84.
- Rolstad B. 2001. The athymic nude rat: An animal experimental model to reveal novel aspects of innate immune responses?. Immunol Rev. 184:136–144.
- Shi Y, Galusha SA, Rock KL. 2006. Cutting edge: Elimination of an endogenous adjuvant reduces the activation of CD8 T lymphocytes to transplanted cells and in an autoimmune diabetes model. J Immunol. 176:3905–3908.
- Srivastava P. 2002. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2:185–194.
- Srivastava PK, Udono H, Blachere NE, Li Z. 1994. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics. 39:93–98.
- Svensson PA, Asea A, Englund MC, Bausero MA, Jernas M, Wiklund O, Ohlsson BG, Carlsson LM, Carlsson B. 2006. Major role of HSP70 as a paracrine inducer of cytokine production in human oxidized LDL treated macrophages. Atherosclerosis. 185:32–38.
- Todryk SM, Melcher AA, Dalgleish AG, Vile RG. 2000. Heat shock proteins refine the danger theory. Immunology. 99:334–337.
- Udelsman R, Blake MJ, Stagg CA, Li DG, Putney DJ, Holbrook NJ. 1993. Vascular heat shock protein expression in response to stress. Endocrine and autonomic regulation of this age-dependent response. J Clin Invest. 91:465–473.
- Udelsman R, Blake MJ, Holbrook NJ. 1991. Molecular response to surgical stress: Specific and simultaneous heat shock protein induction in the adrenal cortex, aorta, and vena cava. Surgery. 110:1125–1131.
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. 2002. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 277:15107–15112.
- van Eden W, Koets A, van Kooten P, Prakken B, van der Zee R. 2003. Immunopotentiating heat shock proteins: Negotiators between innate danger and control of autoimmunity. Vaccine. 21:897–901.
- van Eden W, van der Zee R, Prakken B. 2005. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 5:318–330.
- Velin AK, Ericson AC, Braaf Y, Wallon C, Soderholm JD. 2004. Increased antigen and bacterial uptake in follicle associated epithelium induced by chronic psychological stress in rats. Gut. 53:494–500.
- Walsh RC, Koukoulas I, Garnham A, Moseley PL, Hargreaves M, Febbraio MA. 2001. Exercise increases serum Hsp72 in humans. Cell Stress Chaperones. 6:386–393.
- Wang Y, Seidl T, Whittall T, Babaahmady K, Lehner T. 2010. Stress-activated dendritic cells interact with CD4+T cells to elicit homeostatic memory. Eur J Immunol. 40:1628–1638.
- Wells AD, Rai SK, Salvato MS, Band H, Malkovsky M. 1998. Hsp72-mediated augmentation of MHC class I surface expression and endogenous antigen presentation. Int Immunol. 10:609–617.
- Whitham M, Fortes MB. 2008. Heat shock protein 72: Release and biological significance during exercise. Front Biosci. 13:1328–1339.
- Yokota S, Fujii N. 2010. Immunomodulatory activity of extracellular heat shock proteins and their autoantibodies. Microbiol Immunol. 54:299–307.
- Yoo CG, Lee S, Lee CT, Kim YW, Han SK, Shim YS. 2000. Anti-inflammatory effect of heat shock protein induction is related to stabilization of I kappa B alpha through preventing I kappa B kinase activation in respiratory epithelial cells. J Immunol. 164:5416–5423.
- Zampell JC, Yan A, Avraham T, Andrade V, Malliaris S, Aschen S, Rockson SG, Mehrara BJ. 2011. Temporal and spatial patterns of endogenous danger signal expression after wound healing and in response to lymphedema. Am J Physiol Cell Physiol. 300:C1107–C1121.
- Zedler S, Faist E. 2006. The impact of endogenous triggers on trauma-associated inflammation. Curr Opin Crit Care. 12:595–601.