Abstract
Retrotransposons participate in cellular responses elicited by stress, and DNA methylation plays an important role in retrotransposon silencing and genomic imprinting during mammalian development. Assisted reproduction technologies (ARTs) may be associated with increased stress and risk of epigenetic changes in the conceptus. There are similarities in the nature and regulation of LTR retrotransposons and imprinted genes. Here, we investigated whether the methylation status of Human Endogenous Retroviruses (HERV)-K LTR retrotransposons and the imprinting signatures of the DLK1/MEG3. p57KIP2 and IGF2/H19 gene loci are linked during early human embryogenesis by examining trophoblast samples from ART pregnancies and preimplantation genetic diagnosis (PGD) cases and matched naturally conceived controls. Methylation analysis revealed that HERV-Ks were totally methylated in the majority of controls while, in contrast, an altered pattern was detected in ART-PGD samples that were characterized by a hemi-methylated status. Importantly, DLK1/MEG3 demonstrated disturbed methylation in ART-PGD samples compared to controls and this was associated with altered HERV-K methylation. No differences were detected in p57KIP2 and IGF2/H19 methylation patterns between ART-PGD and naturally conceived controls. Using bioinformatics, we found that while the genome surrounding the p57KIP2 and IGF2/H19 genes differentially methylated regions had low coverage in transposable element (TE) sequences, the respective one of DLK1/MEG3 was characterized by an almost 2-fold higher coverage. Moreover, our analyses revealed the presence of KAP1-binding sites residing within retrotransposon sequences only in the DLK1/MEG3 locus. Our results demonstrate that altered HERV-K methylation in the ART-PGD conceptuses is correlated with abnormal imprinting of the DLK1/MEG3 locus and suggest that TEs may be affecting the establishment of genomic imprinting under stress conditions.
Introduction
The dynamic interrelation between genome and epigenome has a central role in the regulation of gene expression, which is fundamental for normal differentiation during mammalian development. The epigenetic modifications involving, amongst others, DNA methylation and histone modifications can alter cell physiology and homeostasis in response to intrinsic and environmental signals (Jaenisch & Bird, Citation2003). DNA methylation is established during development, maintained in adult somatic cells in a highly orchestrated manner (Szyf, Citation2010) and is essential in several processes, such as X chromosome inactivation, genomic imprinting and retrotransposon silencing (Chen & Riggs, Citation2011).
Genomic imprinting is an important epigenetic regulatory mechanism resulting in monoallelic gene expression related to parental origin (Bartolomei & Tilghman, Citation1997). At present, ∼80 human imprinted genes have been reported, while >150 have been predicted by computational studies (Piedrahita, Citation2011; Wilkins & Úbeda, Citation2011). Of all mechanisms involved in genomic imprinting, DNA methylation is the best studied and is established in CpG-rich domains called differentially methylated regions (DMRs). DNA methylation is catalyzed by DNA methyltransferases (DNMTs), while its maintenance is performed by either DNMTs or trans-acting factors, such as MBD3, PGC7/Stella, ZFP57 and KAP1 (Bartolomei, Citation2009). It should be noted that we used the term “DNA methylation” for covalent modifications of mammalian DNA occurring via the 5-methylation or 5-hydroxymethylation of cytosine typically in the context of the CpG dinucleotide. This is in order to be consistent with primary publications, as other DNA modifications involved in DNA demethylation have been recently identified (Inoue et al., Citation2011). In general, genomic imprints are erased in embryonic germ cells and re-established afterward during gametogenesis and after fertilization (Reik & Walter, Citation2001). Imprinted genes are essential for the correct regulation of both placental and fetal growth (Radford et al., Citation2011; Wilkins & Úbeda, Citation2011) and their epigenetic deregulation can lead to fetal growth abnormalities as well as imprinting-associated changes and disorders (Wilkins & Úbeda, Citation2011).
Transposable elements (TEs) are repetitive genetic elements that constitute over two-thirds of the human genome (de Koning et al., Citation2011). Retrotransposons, which represent the major class of TEs (Cordaux & Batzer, Citation2009), may be mobilized through a RNA-intermediate, which upon its conversion into cDNA by an endogenous reverse transcriptase, is integrated into new genomic sites. They may affect cell functions, which range from local instability to large-scale structural variation, driving genome evolution and altering genetically and/or epigenetically gene expression (Cordaux & Batzer, Citation2009; Goodier & Kazazian, Citation2008). To regulate retrotransposon RNA expression and mobilization activity, cells have developed different defense mechanisms of which DNA methylation is a major one (Goodier & Kazazian, Citation2008). Retrotransposons are generally silenced, as ∼90% of methylated cytosine residues in human DNA lie within retrotransposons (Yoder et al., Citation1997). Their transcriptional activity however is less restrained in proliferating germ and stem cells (Garcia-Perez et al., Citation2007; Georgiou et al., Citation2009), as well as during development and embryogenesis (Kano et al., Citation2009). Retrotransposon activity can be induced by environmental factors and, particularly, stress (Capy et al., Citation2000). Although controlled retrotransposon activity might be beneficial for the cell (Capy et al., Citation2000), the deregulated state may cause monogenic genetic diseases (e.g. haemophilia, cystic fibrosis, Duchenne muscular dystrophy), and has been linked to multifactorial diseases (e.g. cancer, autoimmune diseases) (Cordaux & Batzer, Citation2009; Goodier & Kazazian, Citation2008).
Human Endogenous Retroviruses (HERVs) are long terminal repeat (LTR) retrotransposons that constitute ∼8.3% of the human genome and are derived from germline-integrated proviruses that have undergone endogenization (Bannert & Kurth, Citation2004). HERV-K is the evolutionarily youngest and more active subfamily of HERVs consisting of 91 proviruses, which retained functional full-length open reading frames (ORFs) coding for gag, prt, pol and env, as well as 941 solitary LTRs (Bannert & Kurth, Citation2004; Subramanian et al., Citation2011). HERV-K10, the prototype of HERV-K genome retrotransposon is homologous to the mouse LTR retrotransposon Intracisternal A-Particle (IAP) (Ono et al., Citation1986). The transcriptional activity of HERV-Ks is directly regulated by CpG methylation (Lavie et al., Citation2005). Notably, HERV-K is transcriptionally induced under stress conditions (Cho et al., Citation2008), while its transcripts are detected in human oocytes, lymphocytes and cancer cells (Georgiou et al., Citation2009).
Assisted reproduction technology (ART) involves in vitro manipulation of gametes, such as in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI), and embryo culture and related procedures (Piedrahita, Citation2011). ART accounts for 1–3% of births in developed countries (Shiota & Yamada, Citation2009) and its wide clinical application has led to the development of methods testing for genetic defects, such as preimplantation genetic diagnosis (PGD). ART procedures take place in a critical time-window, during which DNA methylation patterns are initiated, and may be associated with increased parental stress during pregnancy compared to natural conceptions (Kanaka-Gantenbein et al., Citation2010). More specifically, epidemiological data have raised some concern whether ART may increase the risk of epigenetic changes leading to genomic imprinting disorders, either during ART procedures and/or during the resulting pregnancies following ART (Kanaka-Gantenbein et al., Citation2010; Lucifero et al., Citation2004a).
Retrotransposon methylation may influence imprinted genes, as exemplified by the mouse LTR retrotransposon IAP at the Agouti locus (Michaud et al., Citation1994). On the other hand, HERVs respond to stress signals (Cho et al., Citation2008), and stress might cause epigenetic errors in imprinted genes (Jirtle & Skinner, Citation2007). Based on these data, we asked whether LTR retrotransposon methylation status and genomic imprinting signatures are linked following ART-PDG, a condition likely associated with stress (Kanaka-Gantenbein et al., Citation2010). Here, we investigated the methylation status of HERV-Ks – an active HERV subfamily (Bannert & Kurth, Citation2004) – and three well-known imprinted domains DLK1/MEG3. IGF2/H19 and p57KIP2 widely used in methylation analysis studies (Wilkins & Úbeda, Citation2011). Our results showed that following ART-PGD HERV-K methylation changes are correlated with abnormal imprinting of the DLK1/MEG3 locus, which is characterized by a higher coverage in TEs sequences and KAP1-binding sites residing within retrotransposons, compared to the IGF2/H19 and p57KIP2 loci.
Methods
Samples
We used chorionic villi samples, collected in the Laboratory of Medical Genetics (University of Athens), after informed consent of the participants who were undergoing prenatal diagnosis after PGD. The samples were divided in two groups: (i) samples taken after ICSI and PGD (ART-PGD) and (ii) matched samples after natural conceptions, as controls. The confidentiality and anonymity of all participants were ensured by coded references and appropriate safeguards of the data stored. The study was approved by the Ioannina University Hospital Ethics Committee (560/2005).
DNA extraction and bisulfite modification
DNA was extracted from the samples using a commercially available DNA extraction kit (QIAamp DNA extraction Mini Kit, Qiagen, Valencia, CA). Genomic DNA (1–2 μg) derived from each sample was bisulfite modified (Sigma, St. Louis, MO), as previously described (Manning et al., Citation2000). The modification took place at 55 °C in the dark after an overnight (15–17 h) incubation. Subsequently, DNA was purified using the Wizard DNA clean-up system (Promega, Madison, WI), followed by neutralization with ammonium acetate (Sigma) and glycogen (Invitrogen, Carlsbad, CA) and precipitation with 70% ethanol (Sigma). Finally, DNA was diluted in double distilled water and stored at −80 °C until use.
Methylation-specific PCR and statistical analysis
Methylation analysis was performed by methylation-specific PCR (MS-PCR), using specific primers sets for the methylated and unmethylated alleles under conditions shown in . In particular, we used previously described primers for the analysis of DMR methylation status of IGF2/H19 (Poon et al., Citation2002), p57KIP2 (Li et al., Citation2002) and DLK1/MEG3 (Murphy et al., Citation2003) (). For the methylation analysis of HERV-K retrotransposons, we designed primer sets for the methylated and unmethylated allele based on the sequence of HERV-K10 CpG island spanning from 981 to 1085 bp in relation to the transcriptional start site (Ono et al., Citation1986) using MethPrimer software (). HERV-K primer sequences were tested and the exact genomic positions were retrieved using the UCSC in silico PCR tool (Kent et al., Citation2002). All primer sets were designed to amplify the methylated and unmethylated sequences in a way that distinguishes the amplicons by size and the positivity or negativity of the samples on the presence or absence of the PCR products, respectively. As a negative control, reactions without template DNA were performed. MS-PCR products were fractionated in 2% agarose gel and photographed under UV light. All samples were analyzed in triplicate (n = 3). The statistical analysis of MS-PCR experiment data was performed using the chi-squared test. p Values < 0.05 were considered statistically significant.
Figure 1. Primers and conditions used for methylation analysis by MS-PCR. (A) List of primers for the methylated and unmethylated alleles and thermal conditions used for methylation analysis by MS-PCR. MF and MR, forward and reverse primers specific to bisulfite converted methylated DNA; UF and UR, forward and reverse primers specific to bisulfite converted unmethylated DNA; U/M R, reverse primer specific to bisulfite converted unmethylated and methylated DNA. (B) Schematic representation of HERV-K primers designed. In relation to the transcriptional start site of HERV-K10, primers are complementary to the following positions: HERV-K10-UF, 959–983 bp; HERV-K10-MF, 958–983 bp; HERV-K10-UR and HERV-K10-MR, 1036–1060 bp. HERV-K10 NCBI accession number is indicated in parenthesis.

Bioinformatics
The in silico program RepeatMasker (http://www.repeatmasker.org/) was used to measure the percentage coverage of TEs sequences within the DLK1/MEG3. IGF2/H19 and p57KIP2 loci. We used the UCSC genome browser (Kent et al., Citation2002) to extract sequences from the human genome (hg19), encompassing the imprinted gene as well as the genomic region upstream of the DMR of interest and having a total size of ∼126 kb in all three cases. Specifically, the nucleotide sequences analyzed were: (i) chr14:101,201,234–101,327,360 for the DLK1/MEG3 locus, (ii) chr11:2,003,437–2,129,448 for the IGF2/H19 locus and (iii) chr11:2,897,414–3,024,995 for the p57KIP2 locus.
Data mining of KAP1 Chip-seq peaks in DLK1/MEG3. IGF2/H19 and p57KIP2 loci, respectively, was performed using the UCSC table browser (Karolchik et al., Citation2004). Data were extracted from a table with Chip-Seq data from the ENCODE project (wgEncodeRegTfbsClusteredV2), containing, among others, the data sets for KAP1-binding sites (UCSC accession numbers wgEncodeEH001779, wgEncodeEH001776 and wgEncodeEH001779). Retrotransposon-associated KAP1-binding sites were found by intersecting KAP1 Chip-seq peaks and Repeatmasker data tracks in the UCSC table browser. Peaks were viewed and analyzed in the UCSC Genome browser (Kent et al., Citation2002).
Results
Alteration of HERV-K methylation status following ART-PGD compared to natural conceptions
It has been suggested that endogenous retroviruses are permanently inactivated during embryonic development (Rowe & Trono, Citation2011). However, HERV-K methylation status during early human embryogenesis remains unknown so far. To examine this, we investigated the methylation status of HERV-Ks in human trophoblast samples from pregnancies initiated after ICSI and PGD and from matched control samples after natural conceptions, hereafter referred to as PGD and controls, respectively.
To study HERV-K methylation, we designed two sets of primers based on the sequence of CpG island of HERV-K10 (Ono et al., Citation1986) and the specificity of the designed primers was checked using the UCSC in silico PCR tool (Kent et al., Citation2002). Our analysis showed that they specifically amplified HERV-K members located in 17 different chromosomal positions. It should be noted that almost all HERV-Ks amplified were characterized by human-specificity and polymorphisms, while many of them bore full-length ORFs (data not shown), indicating that our primers can detect active HERV-K sequences.
By analyzing control samples, we found that 54 out of 60 samples (90%) exhibited total methylation, while the remaining 6 control samples (10%) presented a pattern of both methylated and unmethylated alleles. In PGD samples, on the other hand, we found that 26 out of 35 samples (74.3%) showed a totally methylated pattern, while 9 out of 35 (25.7%) had both methylated and unmethylated alleles. The differences observed between PGD samples and controls corresponded to a statistically significant alteration of HERV-K methylation (p < 0.042) ().
Table 1. Methylation analysis of HERV-K among PGD and control samples.
These data show that HERV-Ks are usually methylated in trophoblast samples deriving from natural conceptions, while their methylation status is altered following ART-PGD.
Altered HERV-K methylation is correlated with abnormal imprinting of DLK1/MEG3, but not IGF2/H19 and p57KIP2
We next investigated the methylation status of DLK1/MEG3. p57KIP2 and IGF2/H19 imprinted genes in the two groups of samples to address whether genomic imprinting signatures are affected following ART-PGD.
DLK1/MEG3 methylation analysis showed that all control samples exhibited both methylated and unmethylated alleles, as expected. In contrast, we found a statistically significant alteration of DNA methylation pattern in PGD samples (p < 0.001). While 25 out of 35 samples (71.5%) showed the expected pattern of both methylated and unmethylated alleles, 9 samples (25.7%) showed hypomethylation and 1 sample (2.8%) revealed hypermethylation, and hence a disturbed methylation status (Benetatos et al., Citation2010) (). Notably, in 8 out of 10 PGD samples (80%) with abnormal DLK1/MEG3 imprinting, altered methylation of HERV-K was detected as well. Specifically, all these samples (samples AF-139, AF-141, AF-195, AF-210, AF-221, 04-C44, 05-C50 and 05-C61) exhibited hypomethylation of DLK1/MEG3 and a hemi-methylated HERV-K pattern (). As regards p57KIP2 and IGF2/H19, we found that all samples of either the control or the PGD group expressed a pattern of both methylated and unmethylated alleles, thus indicating no difference on the establishment of imprints (, 2C and 3).
Table 2. Methylation analysis of (A) DLK1/MEG3, (B) p57KIP2 and (C) IGF2/H19 DMRs among PGD and control samples.
Table 3. Total results of methylation analyses performed in PGD samples.
Taken together, our data show that altered HERV-K methylation is differentially associated with the methylation status of imprinted loci, correlating with abnormal imprinting of DLK1/MEG3 but having no correlation with that of p57KIP2 and IGF2/H19.
DLK1/MEG3 is characterized by a high coverage in TEs sequences and KAP1-binding sites lying within retrotransposons
The differential methylation status of DLK1/MEG3. p57KIP2 and IGF2/H19 observed prompted us to investigate for other factors that possibly affect the establishment of imprinting signatures. Given that the DMR surrounding genome sequence has a fundamental role in the establishment of genomic imprinting (Reinhart et al., Citation2002), we measured the coverage in TEs sequences within the aforementioned imprinted loci using the in silico program RepeatMasker.
Since MEG3 DMR encompasses nucleotides of both a 90-kb intergenic region and the promoter of MEG3 (Murphy et al., Citation2003), we examined both regions of the imprinted gene cluster DLK1/MEG3. Our analysis revealed that DLK1/MEG3 gene cluster was characterized by coverage in TEs sequences that amounted to 37.37% (). More specifically, we found a significant load in Short Interspersed Nuclear Elements (SINEs) and long interspersed nuclear elements (LINEs) sequences, corresponding to percentage values of 14.05% and 13.16%, respectively. Furthermore, 7.10% of the examined region was covered by LTR retrotransposon and 3.06% by DNA transposon sequences ().
Figure 2. Abundance in transposable element sequences within DLK1/MEG3. IGF2/H19 and p57KIP2 imprinted loci. Histograms indicate the percentage coverage in SINE, LINE, LTR retrotransposon, DNA transposon as well as the total one in transposable element sequences within (A) DLK1/MEG3, (B) IGF2/H19 and (C) p57KIP2 imprinted loci. The tables, below each histogram, show the percentage values of the coverage of SINE, LINE, LTR retrotransposon, DNA transposon and total transposable element sequences within the aforementioned regions.
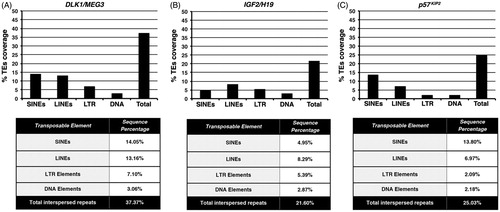
In contrast, respective analyses for p57KIP2 and IGF2/H19 revealed that both loci had a lower content in TEs sequences compared to DLK1/MEG3. Thus, analysis of IGF2/H19 revealed that 21.60% of the nucleotide sequence examined was constituted of TEs. The coverage in SINE, LINE, LTR retrotransposon and DNA transposon sequences was 4.95%, 8.29%, 5.39% and 2.87%, respectively (). Analysis of p57KIP2 revealed an abundance in TEs sequences of 25.03%, almost similar to that of IGF2/H19 (). In p57KIP2, abundance in SINE sequences was slightly lower than DLK1/MEG3 (13.80%). Of note, a significant difference was observed in the coverage in LINE, LTR retrotransposon and DNA transposon sequences, which were significantly lower than the IGF2/H19, with values of 6.97%, 2.09% and 2.18%, respectively.
The different coverage in TEs sequences of the three imprinted loci prompted us to get further insights into their possible contribution to the imprinting changes previously observed. Given that KAP1 is essential for DNA methylation of endogenous retroviruses (ERVs) (Rowe et al., Citation2013) and possibly involved in the establishment of imprinting during embryogenesis (Strogantsev & Ferguson-Smith, Citation2012), we examined DLK1/MEG3. p57KIP2 and IGF2/H19 imprinted loci for presence of KAP1-binding sites. Bioinformatic analysis of the DLK1/MEG3 locus revealed the existence of four KAP1-binding sites. Interestingly, three out of four sites resided within retrotransposon sequences. Two of them (site 1 and 2) were located at the boundary of two neighboring LINE and LTR retrotransposons and composed by part of their sequences. More precisely, site 1 (chr14: 101,222,006–101,222,455) bore sequences of the MLT1A0 member of LTR retrotransposon ERVL-MaLR family and the L1MB8 LINE, while site 2 (chr14: 101,228,449–101,228,898) those of the L1MB8 LINE and the ERV3-16A3_I-int ERV-L LTR retrotransposon. Site 3 (chr14: 101,274,622–101,275,071) was partially composed by sequences of the FLAM_A member of the Alu family of SINE retrotransposons (). Respective analyses of the p57KIP2 and IGF2/H19 loci revealed one KAP1-binding site not residing within retrotransposon sequences in the former () and absence of KAP1-binding sites in the latter (), respectively.
Figure 3. Bioinformatic analysis for KAP1-binding sites within DLK1/MEG3. p57KIP2 and IGF2/H19 loci. KAP1-binding sites were viewed and analyzed in the UCSC Genome Browser. (A) The snapshot, in the upper part, shows in the first track the data from the analysis for KAP1-binding sites within DLK1/MEG3 locus. KAP1-all denotes the total KAP1-binding sites found, while KAP1 the retrotransposon-associated KAP1-binding sites, respectively. The next two tracks show the genes and the CpG islands contained in the genomic region, respectively. The Repeating Elements cluster shown in the last track summarizes the set of transposable element sequences from RepeatMasker. In the lower part, the snapshots, named site 1, 2 and 3, indicate the individual retrotransposon-associated KAP1-binding sites within DLK1/MEG3 locus. (B, C) The snapshots show the data from the respective analyses for p57KIP2 and IGF2/H19 loci, respectively.

Collectively, these results show that while the abundance of TEs in p57KIP2 and IGF2/H19 loci is low, the DLK1/MEG3 locus is characterized by a higher coverage in TEs sequences and the presence of KAP1-binding sites lying within retrotransposons.
Discussion
This study provides experimental evidence for a correlation between altered HERV-K methylation and abnormal imprinting of the DLK1/MEG3 locus in human trophoblast samples from ART-PGD compared to normal conceptions. The correlation between HERV-K LTR retrotransposon and imprinted gene methylation status under stress conditions may uncover a possible role of TEs in the establishment of a normal imprinting pattern of certain imprinted genes, such as DLK1/MEG3.
We showed that HERV-K retrotransposons are usually totally methylated during early human embryonic development. Our results are in line with the current viewpoint, based on studies performed solely in mice (Rowe & Trono, Citation2011). Unexpectedly, we found a number of samples in the control group (10%) revealing both methylated and unmethylated alleles. This could be attributed to possible inter-individual variations of LTR DNA methylation, as already documented for the mouse LTR retrotransposon IAP (Waterland & Jirtle, Citation2003). Thus, the hemi-methylated pattern of HERV-K in natural conception samples might be due to the effect of intrinsic or extrinsic/environmental stress and/or other hitherto unknown factors, which lead to a relaxation of epigenetic control during early pregnancy.
One key finding was the significant alteration of HERV-K methylation status following ART-PGD, documented by a hemi-methylated pattern. This epigenetic perturbation could be due to stress elicited during ART-PGD as the result of distinct events occurring during the preimplantation and/or postimplantation stages. First, an altered gene expression has been reported in human preimplantation embryos attributed to stress conditions, with Rb being one of the affected genes (Wells et al., Citation2005). Notably, a correlation between Rb pathway and HERV-K expression has been reported (Li et al., Citation2010). Thus, HERV-K epigenetic deregulation might be the result of Rb altered expression. Second, reactive oxygen species in ART culture media can potentially activate HERV-K, since it has been suggested that oxidative stress might establish a hypomethylation status of certain endogenous retrovirus loci (Cho et al., Citation2008). Third, ovarian hyperstimulation may lead to production of supraphysiological serum estradiol levels (Santos et al., Citation2010), known to activate HERV-K (Ono et al., Citation1987). Fourth, it cannot be excluded that ART, as an extended exposure to the in vitro environment, and PGD, as an invasive method (Georgiou et al., Citation2006), may lead to epigenetic relaxation during preimplantation and/or postimplantation embryonic development.
The other interesting finding of our study was the observation of a differential methylation status between DLK1/MEG3, and p57KIP2 or IGF2/H19 following ART-PGD. While the imprinting status of p57KIP2 and IGF2/H19 was not disturbed in the PGD group, in agreement with other results in IVF and ICSI samples (Tierling et al., Citation2010), we found a significant alteration of DLK1/MEG3 methylation. It has been suggested that two fundamental cis-acting elements are required for the establishment of an imprint on a gene: (i) a DMR, which is necessary but not sufficient alone, and (ii) the surrounding genome sequence (Reinhart et al., Citation2002). In view of the differential methylation observed, we were challenged to evaluate genomic features of the imprinted loci studied, such as the load in TEs sequences of their DMR surrounding genome. Strikingly, our findings corroborated the above suggestion since following ART-PGD a disturbed methylation status was only detected in an imprinted gene characterized by a high coverage in TEs sequences in its DMR surrounding genome. Remarkably, DLK1/MEG3 was characterized by a higher load in sequences of all the TE families and, in total, ∼1.5- and 1.7-fold higher coverage than those of p57KIP2 and IGF2/H19, respectively. It should be mentioned that these findings are combatible with a previous study showing that SINE depletion and LINE abundance at imprinted loci are not features universally required for imprinting (Cowley et al., Citation2011). Moreover, another feature that differed between the imprinted loci studied was the presence of KAP1-binding sites located within retrotransposons solely in DLK1/MEG3, the only locus characterized by disturbed imprinting. Important recent work documented that KAP1 shapes DNA methylation at ERV-containing loci in early development (Rowe et al., Citation2013). Therefore, we suggest that the abundance in TEs sequences of the imprinted gene DMR surrounding genome, as well as the presence of KAP1-binding sites lying within those elements, may represent locus-specific features affecting the establishment of DNA methylation under stress.
We found that the vast majority of PGD samples with abnormal imprinting of DLK1/MEG3 was accompanied by a disturbed methylation pattern of HERV-K. Two lines of evidence agree with our results. First, it was previously proposed that the epigenetic regulation of LTR retrotransposons and imprinted genes shares similarity due to the repeat-like nature of the imprinted gene DMRs (Lucifero et al., Citation2004b). Second, it was documented that DMR-associated genomic imprinting in mammals can originate from the repression of retrotransposons by DNA methylation (Suzuki et al., Citation2007). Based on these, it is tempting to propose that the regulation of HERV-K retrotransposon methylation and genomic imprinting of loci with certain features of the DMR surrounding genome might be common under stress conditions during early human embryogenesis.
ART procedures may be associated with increased stress compared with spontaneous conception and take place in a time frame critical for the re-establishment of DNA methylation patterns (Kanaka-Gantenbein et al., Citation2010). The functional consequences of the differential methylation observed at HERV-Ks are unknown so far. The imprinting defects of DLK1/MEG3 have been associated with uniparental disomy of chromosome 14 (Murphy et al., Citation2003) and Prader–Willi syndrome-like phenotype (Hosoki et al., Citation2009) as well as several types of cancer (Benetatos et al., Citation2010, Citation2013). However, MEG3 function remains poorly understood (Benetatos et al., Citation2013). Given that ART may be linked to epigenetic risks as well as having some impact on the health of the offspring in later adult life (Kanaka-Gantenbein et al., Citation2010), the differential methylation observed might have long-term effects. HERVs are affected by stress signals and their deregulation has been associated with multifactorial diseases (Cho et al., Citation2008; Cordaux & Batzer, Citation2009; Goodier & Kazazian, Citation2008). Moreover, imprinted genes are possible targets of disease-causing epigenetic errors induced by stress (Jirtle & Skinner, Citation2007). It is widely accepted that stress-related DNA methylation alterations leading to human diseases might involve changes in networks of genes (Szyf, Citation2009). In our opinion, HERVs could be important components of such networks regulating gene expression. Following early life stress, the disturbed methylation of HERVs might serve as mark of a life-long lasting epigenetic “memory”. Upon stress stimuli in adult life, this “memory” can be reactivated leading to HERV deregulation, further responsible for: (i) the induction of genome instability and (ii) alterations in gene expression and, particularly, that of imprinted genes. Thus, HERVs and imprinted genes might participate in a stress-driven gene network, involved in the transgenerational inheritance of stress and the pathogenesis of multifactorial diseases.
In the present work, by conducting a qualitative analysis, we provide evidence of a correlation between HERV-K and imprinted gene methylation status following ART-PGD. A limitation of our study is the lack of direct nucleotide sequencing of the MS-PCR results. Future studies will shed more light in the factors conferring epigenetic lability in response to stress during human embryogenesis and future quantitative analyses are essential to determine the methylation profile of retrotransposons and imprinted genes and whether stress-induced changes in methylation are associated with different mRNA expression patterns in the early life stages. In addition, functional studies may help to establish whether epigenetic modifications of KAP1-binding sites within the DMR surrounding genome have a direct effect on KAP1 binding and imprinted gene expression. Finally, recent technological advances can provide the required data to map long-range intra- and inter-chromosomal interactions and gain insights into the locus-specific features that are important for the establishment of genomic imprinting.
Overall, our results document a correlation between altered HERV-K methylation and abnormal imprinting under conditions likely associated with stress during early human embryogenesis. Further studies will reveal the interwoven structural and functional roles of TEs in the establishment of genomic imprinting.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article. This work was supported by institutional funds of University of Ioannina.
Acknowledgements
The authors thank the patients for their participation in the study. We also thank Prof. Ioannis Georgiou (Genetics and IVF Unit, Department of Obstetrics and Gynecology, Medical School, University of Ioannina, Ioannina, Greece) for the critical reviewing of the manuscript.
Related Research Data
References
- Bannert N, Kurth R. (2004). Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci USA 101(Suppl. 2):14572–9
- Bartolomei MS. (2009). Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev 23(18):2124–33
- Bartolomei MS, Tilghman SM. (1997). Genomic imprinting in mammals. Annu Rev Genet 31:493–525
- Benetatos L, Hatzimichael E, Dasoula A, Dranitsaris G, Tsiara S, Syrrou M, Georgiou I, Bourantas KL. (2010). CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res 34(2):148–53
- Benetatos L, Hatzimichael E, Londin E, Vartholomatos G, Loher P, Rigoutsos I, Briasoulis E. (2013). The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci 70(5):795–814
- Capy P, Gasperi G, Biémont C, Bazin C. (2000). Stress and transposable elements: co-evolution or useful parasites? Heredity (Edinb) 85(Pt 2):101–6
- Chen ZX, Riggs AD. (2011). DNA methylation and demethylation in mammals. J Biol Chem 286(21):18347–53
- Cho K, Lee YK, Greenhalgh DG. (2008). Endogenous retroviruses in systemic response to stress signals. Shock 30(2):105–16
- Cordaux R, Batzer MA. (2009). The impact of retrotransposons on human genome evolution. Nat Rev Genet 10(10):691–703
- Cowley M, de Burca A, McCole RB, Chahal M, Saadat G, Oakey RJ, Schulz R. (2011). Short interspersed element (SINE) depletion and long interspersed element (LINE) abundance are not features universally required for imprinting. PLoS One 6(4):e18953
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7(12):e1002384
- Garcia-Perez JL, Marchetto MC, Muotri AR, Coufal NG, Gage FH, O’Shea KS, Moran JV. (2007). LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet 16(13):1569–77
- Georgiou I, Noutsopoulos D, Dimitriadou E, Markopoulos G, Apergi A, Lazaros L, Vaxevanoglou T, et al. (2009). Retrotransposon RNA expression and evidence for retrotransposition events in human oocytes. Hum Mol Genet 18(7):1221–8
- Georgiou I, Syrrou M, Pardalidis N, Karakitsios K, Mantzavinos T, Giotitsas N, Loutradis D, et al. (2006). Genetic and epigenetic risks of intracytoplasmic sperm injection method. Asian J Androl 8(6):643–73
- Goodier JL, Kazazian HH. (2008). Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135(1):23–35
- Hosoki K, Kagami M, Tanaka T, Kubota M, Kurosawa K, Kato M, Uetake K, et al. (2009). Maternal uniparental disomy 14 syndrome demonstrates Prader--Willi syndrome-like phenotype. J Pediatr 155(6):900–3.e1
- Inoue A, Shen L, Dai Q, He C, Zhang Y. (2011). Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res 21(12):1670–6
- Jaenisch R, Bird A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–54
- Jirtle RL, Skinner MK. (2007). Environmental epigenomics and disease susceptibility. Nat Rev Genet 8(4):253–62
- Kanaka-Gantenbein C, Sakka S, Chrousos GP. (2010). Assisted reproduction and its neuroendocrine impact on the offspring. Prog Brain Res 182:161–74
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH Jr. (2009). L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23(11):1303–12
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32(Database issue):D493–6
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. (2002). The human genome browser at UCSC. Genome Res 12(6):996–1006
- Lavie L, Kitova M, Maldener E, Meese E, Mayer J. (2005). CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J Virol 79(2):876–83
- Li Y, Nagai H, Ohno T, Yuge M, Hatano S, Ito E, Mori N, et al. (2002). Aberrant DNA methylation p57(KIP2) gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood 100(7):2572–7
- Li Z, Sheng T, Wan X, Liu T, Wu H, Dong J. (2010). Expression of HERV-K correlates with status of MEK-ERK and p16INK4A-CDK4 pathways in melanoma cells. Cancer Invest 28(10):1031–7
- Lucifero D, Chaillet JR, Trasler JM. (2004a). Potential significance of genomic imprinting defects for reproduction and assisted reproductive technology. Hum Reprod Update 10(1):3–18
- Lucifero D, Mann MR, Bartolomei MS, Trasler JM. (2004b). Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet 13(8):839–49
- Manning M, Lissens W, Bonduelle M, Camus M, De Rijcke M, Libaers I, Van Steirteghem A. (2000). Study of DNA methylation patterns at chromosome 15q11-q13 in children born after ICSI reveals no imprinting defects. Mol Hum Reprod 6(11):1049–53
- Michaud EJ, van Vugt MJ, Bultman SJ, Sweet HO, Davisson MT, Woychik RP. (1994). Differential expression of a new dominant agouti allele (Aiapy) is correlated with methylation state and is influenced by parental lineage. Genes Dev 8(12):1463–72
- Murphy SK, Wylie AA, Coveler KJ, Cotter PD, Papenhausen PR, Sutton VR, Shaffer LG, Jirtle RL. (2003). Epigenetic detection of human chromosome 14 uniparental disomy. Hum Mutat 22(1):92–7
- Ono M, Kawakami M, Ushikubo H. (1987). Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. J Virol 61(6):2059–62
- Ono M, Yasunaga T, Miyata T, Ushikubo H. (1986). Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J Virol 60(2):589–98
- Piedrahita JA. (2011). The role of imprinted genes in fetal growth abnormalities. Birth Defects Res A Clin Mol Teratol 91(8):682–92
- Poon LL, Leung TN, Lau TK, Chow KC, Lo YM. (2002). Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem 48(1):35–41
- Radford EJ, Ferrón SR, Ferguson-Smith AC. (2011). Genomic imprinting as an adaptative model of developmental plasticity. FEBS Lett 585(13):2059–66
- Reik W, Walter J. (2001). Genomic imprinting: parental influence on the genome. Nat Rev Genet 2(1):21–32
- Reinhart B, Eljanne M, Chaillet JR. (2002). Shared role for differentially methylated domains of imprinted genes. Mol Cell Biol 22(7):2089–98
- Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, Aktas T, Trono D. (2013). De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 140(3):519–29
- Rowe HM, Trono D. (2011). Dynamic control of endogenous retroviruses during development. Virology 411(2):273–87
- Santos MA, Kuijk EW, Macklon NS. (2010). The impact of ovarian stimulation for IVF on the developing embryo. Reproduction 139(1):23–34
- Shiota K, Yamada S. (2009). Intrauterine environment--genome interaction and children’s development (3): assisted reproductive technologies and developmental disorders. J Toxicol Sci 34(Suppl. 2):SP287–91
- Strogantsev R, Ferguson-Smith AC. (2012). Proteins involved in establishment and maintenance of imprinted methylation marks. Brief Funct Genomics 11(3):227–39
- Subramanian RP, Wildschutte JH, Russo C, Coffin JM. (2011). Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90
- Suzuki S, Ono R, Narita T, Pask AJ, Shaw G, Wang C, Kohda T, et al. (2007). Retrotransposon silencing by DNA methylation can drive mammalian genomic imprinting. PLoS Genet 3(4):e55
- Szyf M. (2009). Early life, the epigenome and human health. Acta Paediatr 98(7):1082–4
- Szyf M. (2010). DNA methylation and demethylation probed by small molecules. Biochim Biophys Acta 1799(10–12):750–9
- Tierling S, Souren NY, Gries J, Loporto C, Groth M, Lutsik P, Neitzel H, et al. (2010). Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J Med Genet 47(6):371–6
- Waterland RA, Jirtle RL. (2003). Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23(15):5293–300
- Wells D, Bermúdez MG, Steuerwald N, Malter HE, Thornhill AR, Cohen J. (2005). Association of abnormal morphology and altered gene expression in human preimplantation embryos. Fertil Steril 84(2):343–55
- Wilkins JF, Úbeda F. (2011). Diseases associated with genomic imprinting. Prog Mol Biol Transl Sci 101:401–45
- Yoder JA, Walsh CP, Bestor TH. (1997). Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13(8):335–40