
Abstract
Stress is known to correlate with the incidence of acute myocardial infarction. However, the molecular mechanisms underlying this correlation are not known. This study was designed to assess the effect of experimental stress on arterial thrombus formation, the key event in acute myocardial infarction. Mice exposed to 20 h of restraint stress displayed an increased arterial prothrombotic potential as assessed by photochemical injury-induced time to thrombotic occlusion. This increase was prevented by chemical sympathectomy performed through 6-hydroxydopamine (6-OHDA). Blood-born tissue factor (TF) activity was enhanced by stress and this increase could be prevented by 6-OHDA treatment. Vessel wall TF, platelet count, platelet aggregation, coagulation times (PT, aPTT), fibrinolytic system (t-PA and PAI-1) and tail bleeding time remained unaltered. Telemetric analysis revealed only minor hemodynamic changes throughout the stress protocol. Plasma catecholamines remained unaffected after restraint stress. Tumor necrosis factor alpha (TNF-α) plasma levels were unchanged and inhibition of TNF-α had no effect on stress-enhanced thrombosis. These results indicate that restraint stress enhances arterial thrombosis via the sympathetic nervous system. Blood-borne TF contributes, at least in part, to the observed effect whereas vessel wall TF, platelets, circulating coagulation factors, fibrinolysis and inflammation do not appear to play a role. These findings shed new light on the understanding of stress-induced cardiovascular events.
Introduction
In modern society, people are frequently faced with stressful situations. Several lines of evidence (Rosengren et al., Citation2004) demonstrate a correlation between stress and cardiovascular events. However, the mechanisms accounting for these stress-related incidents are not well understood.
The sympathetic nervous system (SNS) is one of the most intensely studied pathways mediating responses to stress and accounts for the increased vascular morbidity and mortality in response to several stressors (Nawrot et al., Citation2011). Arterial thrombus formation is the critical step in acute cardiovascular events such as myocardial infarction (Ross, Citation1999), and tissue factor (TF) is a key trigger of thrombus formation in vivo (Steffel et al., Citation2006). In addition, inflammation and in particular cytokines such as tumor necrosis factor alpha (TNF-α) (Yamamoto et al., Citation2002) have been proposed as potential mediators of stress related vascular events, as have other components of the coagulation cascade (as well as platelets) (Larsson et al., Citation1989).
Restraint stress is a long established model of experimental stress for rodents (Glavin et al., Citation1994) and allows for the characterization of the mechanisms involved (Yamamoto et al., Citation2002). To that end, we measured in vivo real-time arterial thrombus formation (Stampfli et al., Citation2010) in stressed mice. To assess the role of the SNS, we used 6-hydroxydopamine (6-OHDA), a highly specific blocker of the sympathetic nervous system. This procedure, known as chemical sympathectomy (Finch et al., Citation1972), leads to destruction of the post-synaptic sympathetic neurons. Since TNF-α has been proposed as a potential mediator of stress related vascular events (Yamamoto et al., Citation2002), the role of TNF-α was assessed using etanercept, a highly specific soluble blocking receptor for TNF-α, which has been proven to reliably inhibit TNF-α in vivo in C57Bl/6 mice (Hui-Yuen et al., Citation2006).
Methods
Animals
Age-matched male C57Bl/6 mice were randomly assigned to control conditions or restraint stress. Mice were bred and housed in a specific pathogen free (SPF) animal facility with conventional light cycle at the Institute of Laboratory Animal Science, University of Zurich. All experiments were approved by the Veterinary Office and the Department of Health of the Canton of Zurich, Switzerland.
Stress protocol
Mice were placed into 50-ml conical centrifuge tubes fitted with multiple punctures allowing ventilation and water uptake. Tubes were placed in horizontal holders inside a standard cage housed in the animal facility with conventional light cycle. Water was provided ad libitum to the animals during the entire procedure. After 20 h of restraint stress (Yamamoto et al., Citation2002), mice were anesthetized without delay and exposed to the arterial thrombosis protocol (Stampfli et al., Citation2010). After cessation of the experiment, animals were euthanized by pentobarbital overdose injection and relevant organs were excised. All groups were food deprived 20 h prior to the thrombosis experiment.
Arterial thrombosis
A separate set of mice was anesthetized with Pentobarbital (87 mg/kg i.p.). Rose Bengal (Fisher Scientific, Fair Lawn, NJ) was injected into the tail vein in a volume of 0.12 ml at a concentration of 50 mg/kg body weight. Mice were placed under a dissecting microscope and the right common carotid artery was exposed following a midline cervical incision. A Doppler flow probe (Model 0.5 VB, Transonic Systems, Ithaca, NY) was applied and connected to a flowmeter (Transonic, Model T106) supplying a data acquisition system (PowerLab 4/30, AD Instruments, Colorado Springs, CO). A 1.5-mW green light laser (540 nm) (Melles Griot, Carlsbad, CA) was directed at the desired site of injury at a distance of 6 cm for 60 min or until complete occlusion. Flow was monitored real-time for 120 min from the onset of injury. Occlusion was defined as flow ≤0.1 ml/min for at least 1 min (Camici et al., Citation2006). Data were analyzed with ChartPro Software (AD Instruments). The carotid arteries of these mice were isolated for histologic assessment.
6-OHDA treatment
6-OHDA treatment was performed with 6-hydroxydopamine hydrobromide (Sigma-Aldrich, Buchs, Switzerland) as previously described (Kohm & Sanders, Citation1999). 6-OHDA was dissolved in 0.5 M saline containing 1 mM ascorbate as an antioxidant. Mice were treated by i.p. injections (200 mg/kg body weight) 6, 4 and 2 days before the stress protocol. In control groups, vehicle injections (0.5 M saline containing 1 mM ascorbate without 6-OHDA) were performed at the same time points with the same volume as for 6-OHDA treated mice.
Telemetric assessment of blood pressure and heart rate
A separate set of mice was used for this experiment. After anesthesia with xylazine and ketamine, shaving and disinfecting the neck, the left common carotid artery was exposed. The transmitter’s catheter was inserted into the artery and pushed forward until the tip just entered the thoracic aorta. The transmitter body was fixed under the skin. Blood pressure transmitter implantation was carried out under aseptic conditions. Mice were allowed to recover for 2 weeks. Measurements were performed using a TA11PA-C10 transmitter (Data Sciences International, Tilburg, The Netherlands). After implantation and recovery period, parameters were measured telemetrically (without any additional handling) in all groups. After termination of 20 h measurement, mice were euthanized by pentobarbital overdose. Data were generated and analyzed by using Dataquest A.R.T 3.0 software (Data Sciences International, Tilburg, The Netherlands). Organs of these mice were not harvested for further analysis.
Catecholamine measurement
Mice were taken out of their cage or the restraint tube, immediately anesthetized with Pentobarbital (87 mg/kg i.p.), and put in a separate cage for 2 min to allow full effect of the drug. Then, intracardial blood was drawn without any further manipulation. Citrated blood samples were centrifuged at 3000 rpm for 10 min at 4 °C. Subsequently, plasma was collected and quickly frozen at −80 °C for a maximum of 7 days until catecholamine measurements were performed by HPLC with amperometric detection: plasma (120 μl) or standard with dihydroxybenzylamine (DHBA, Sigma Aldrich, Frankfurt, Germany) as internal standard was extracted on activated alumina at pH 8.6. The alumina was allowed to settle and the supernatant aspirated followed by three washes with water. The catecholamines were then eluted with 130 μl of a mixture of acetic acid 0.2 M and phosphoric acid 0.04 M (8/2, v/v) and 100 μl was injected into the chromatography system. The separation was achieved on a reversed-phase column Nucleosil 5 μm C-18, 25 cm × 4.6 mm (Macherey-Nagel AG, Oensingen, Switzerland) using a 50 mM sodium acetate buffer mobile phase containing 20 mM citric acid, 0.135 mM EDTA, 1 mM dibutylamine and 3.8 mM sodium octyl sulfonate, as an ion-pairing agent, and 7% methanol at a flow rate of 0.7 ml/min. The electrochemical detector (from Antec, model Decade) was set at +0.8 V. The following order of elution was observed: noradrenaline (NA), adrenaline (A), DHBA and dopamine. The recovery was 80% and the quantification limit was 5 pg per injection. The interassay coefficients of variation were 11% for NA and 12% for A.
Tissue factor activity
Carotid arteries were homogenized in 50 µl of lysis buffer (50 mM Tris–HCl, 100 mM NaCl, 0.1% Triton X-100, pH 7.4) by manual grinding on ice. Samples were then centrifuged at 14,000 rpm for 15 min at 4 °C and the supernatant was transferred to a fresh tube. For blood-borne TF activity, citrated blood (3.2% citrate 1/10) was harvested by puncture of the right ventricle. Plasma was extracted by 15 min centrifugation at 2500 G and stored immediately at −80 °C until analysis. TF activity was assessed with a colorimetric assay (Actichrome TF, American Diagnostica, Stamford, CT); incubation at 37 °C allowed TF to form a complex with FVII and this complex to cleave FX to FXa. Absorbance was determined at 405 nm and compared to a standard curve generated using known amounts of lyophilized TF (Stampfli et al., Citation2010).
Platelet aggregation
Platelet aggregation was studied in fresh citrated blood within 1 h using a Chrono-Log whole blood impedance aggregometer (Chrono-Log, Havertown, PA). Platelets were equilibrated under constant stirring for 1 min prior to addition of thrombin (0.5 U/ml; Sigma Aldrich, Frankfurt, Germany). Aggregation was displayed as a function of time (AGGRO/LINK Software; Chrono-Log).
Tail bleeding time
Tail bleeding time was assessed as described previously (Renne et al., Citation2005). After anesthetizing mice with Pentobarbital (87 mg/kg body weight, i.p.), the distal 3-mm segment of the tail was removed with a scalpel. Bleeding was monitored by gently absorbing the blood with a filter paper at 15 s intervals without touching the wound.
Platelet count
Platelets were counted in fresh whole blood collected in EDTA tubes using an automated cell counter ScilVet abc plus with impedance technology (Horiba Instruments, Darmstadt, Germany).
PT and aPTT
Prothrombin time (PT) and partial thromboplastin time (aPTT) measurements were conducted by using the Start4 analyzer (Diagnostica Stago, France) with supplied reagents (Roche Diagnostics, Switzerland). Briefly, PT assessed fibrin formation in plasma triggered by the addition of exogenous TF, thus testing the coagulation factors downstream of TF. aPTT also assessed fibrin formation, but this time triggered by the addition of kaolin, which led to contact activation of the intrinsic pathway. In both tests, calcium was added to the plasma.
Plasminogen activator inhibitor ELISA
Plasminogen activator inhibitor (PAI) 1 antigen was determined in citrated plasma using a PAI-1 functional assay ELISA kit (Oxford Biomedical Research, Oxford, MI) according to the manufacturer’s instructions.
Real-time PCR
Carotid arteries were snap frozen in liquid nitrogen and stored at −80 °C until use. Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). cDNA was generated using Ready-To-Go You-Prime First-Strand Beads (Amersham Biosciences, Piscataway, NJ) and first-strand cDNA primer pd(N)6. Real-time PCR was performed using SybrGreen Jump start kit (Sigma). Murine primers were as follows: TF: forward (5-3: CAATGAATTCTCGATTGATGTGG) and reverse (5-3: GGAGGATGATAAAGATGGTGGC). Tissue plasminogen activator (tPA): forward (5-3: TGAGCCAACGCAGACAACTTA) and reverse (5-3: TGACAGCACCCAGCAGGAACT). Data were normalized to murine S12: forward (5-3: GAAGCTGCCAAAGCCTTAGA) and reverse (5-3: AACTGCAACCAAAACCCTTC). The amplification program consisted of 1 cycle at 95 °C for 10 min, followed by 40 cycles with a denaturing phase at 95 °C for 30 s, an annealing phase of 1 min at 60 °C and an elongation phase at 72 °C for 1 min. For verification of the correct amplification, PCR products were analyzed on an ethidium bromide stained 1.5% agarose gel. Quantification was performed using 2 -ΔΔCt method.
Etanercept treatment
Etanercept (Enbrel®; Amgen, Switzerland) was dissolved in the supplied vehicle according to the manufacturer’s instructions and diluted in sterile PBS. Mice were given an i.p. dose of 10 mg/kg 24 h (Hui-Yuen et al., Citation2006) prior to the start of the stress protocol. Control mice were injected with the same amount of vehicle and PBS at the same time.
Immunohistochemistry
Carotid arteries were mounted at optimal cutting temperature (Tissue-Tek). Specimens were cut into 8 -μm sections and fixed with 4% paraformaldehyde. Sections were stained with oil red O for 1 h at room temperature. For the assessment of CD68 (Serotec; primary antibody dilution 1:400), CD3 (Serotec; primary antibody 1:100), VCAM-1 (Serotec; primary antibody 1:200), E-selectin (BD Pharmingen; primary antibody 1:25) and P-selectin (BD Pharmingen; primary antibody 1:50) sections were fixed in acetone, blocked with 1% FCS and incubated with the specific rat anti-mouse primary antibody. The primary antibodies were revealed by goat anti-rat immunoglobulin antibodies (Caltag Laboratories; secondary antibody 1:150), followed by alkaline phosphatase-labeled donkey anti-goat antibodies (Jackson Immunoresearch; dilution 1:80). Alkaline phosphatase was visualized using naphthol AS-BI phosphate and new fuchsin as substrate. Sections were counterstained with hemalum and coverslipped. Atherosclerotic plaques served as positive control and primary murine IgG1 antibody as negative control. From each specimen, six sections in a distance of 100 μm from each other were analyzed. CD68 positive cells in the intimal and medial layer were counted in each cross-section and mean numbers were calculated for each specimen under blind conditions. For VCAM-1, percentages of stained area were quantified with the AnalysisFIVE program.
Statistical analysis
Data are represented as mean ± SEM. Statistical analysis was performed by 2-tailed unpaired Student’s t test or two-way ANOVA as appropriate. A probability value of <0.05 was considered significant.
Results
Time to thrombotic occlusion of the carotid artery was greatly decreased in stressed mice as compared to non-stressed controls (n = 6, p < 0.05, , left), whereas initial carotid blood flow was comparable in both groups (, middle). Chemical sympathectomy, attained by 6-OHDA treatment, abrogated the pro-thrombotic effect of stress as compared to vehicle treated stressed mice [F(1,20) = 22.6, p < 0.01, , right].
Figure 1. (A) Time to thrombotic occlusion of the carotid artery is decreased in mice after restraint stress (n = 6, p < 0.05, left). Initial carotid blood flow is comparable in both groups (middle). Chemical sympathectomy (6-OHDA treatment) blocks the effect of restraint stress on thrombotic occlusion (n = 5, p = n.s., right). (B) TF activity in plasma is increased in stressed mice as compared to controls (n = 5, p < 0.05, left). This increase is prevented by 6-OHDA treatment (n = 5, p = n.s., left). TF activity in the vessel wall is similar in stressed mice and controls irrespective of chemical sympathectomy (n = 5, p = n.s., right). (C) Platelet aggregation to thrombin remains unaltered by stress with or without chemical sympathectomy (n = 5, p = n.s., left). Tail bleeding time (n = 5, p = 0.57, middle) as well as plasma activity of the extrinsic (PT) and intrinsic (aPTT) coagulation pathway are unaffected by stress (n = 4–5, p = 0.73 and 0.48, respectively, right). *p < 0.05; **p < 0.01.
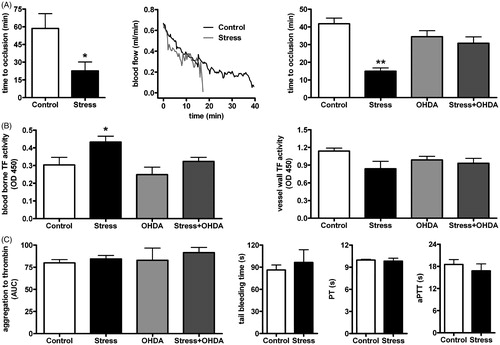
Blood-borne TF activity was increased in plasma of stressed mice as compared to non-stressed controls [F(1,16) = 7.74, p < 0.05] and this increase was prevented by 6-OHDA treatment [F(1,16) = 4.99, p = n.s., , left]. In contrast, TF activity (n = 5, p = n.s., , right) and expression (n = 5, p = n.s., data not shown) in the vessel wall were similar in stressed mice and controls and remained unaffected by 6-OHDA treatment.
Platelet aggregation to thrombin remained unaffected by stress with or without 6-OHDA treatment (n = 5, p = n.s., , left). Similarly, platelet count was not affected by stress with or without 6-OHDA treatment (n = 5, p = n.s., data not shown). Consistent with these observations, tail bleeding time was not affected by stress (n = 5, p = 0.57, , middle).
Activity of extrinsic and intrinsic coagulation pathway (PT and aPTT, respectively) exhibited no difference in plasma of stressed mice versus controls (PT: n = 4–5, p = 0.73; aPTT: n = 4–5, p = 0.48, , right). The fibrinolytic system was examined by tPA expression in arterial homogenates and PAI-1 levels in plasma. Both tPA [0.16 ± 0.03 versus 0.11 ± 0.02 tPA/S12, n = 5, F(1,16) = 0.29, p = 0.20] and PAI-1 [0.106 ± 0.03 versus 0.071 ± 0.02 OD450, n = 5, F(1,16) = 8.39, p = 0.36] remained unaffected by restraint stress.
Blood pressure and heart rate were measured telemetrically. Within the first hour of stress, a moderate increase in mean arterial blood pressure was observed in stressed as compared to control mice after 60 min [n = 7–8, F(1,26) = 8.23, p < 0.05, ]. In 6-OHDA treated mice, a similar, non-significant trend was observed in stressed mice as compared to the respective control mice. Values normalized after 1 h resulting in a similar area under the curve in all the groups for systolic, diastolic and mean arterial pressure as well as heart rate (n = 7–8, p = n.s., ).
Figure 2. (A) Area under the curve is comparable in all the groups (n = 7–8) for systolic, diastolic and mean arterial blood pressure as well as heart rate. Full lines represent mean values of diastolic, mean and systolic blood pressure values; dashed lines represent mean heart rates. (B) Mean arterial blood pressure was elevated in stressed as compared to control mice after 60 min (n = 7–8, p < 0.05). (C) 6-OHDA treatment induced lower plasma levels of noradrenaline in control mice and higher levels of adrenaline in stressed mice. Stress itself had no significant effect on catecholamine levels (n = 5). (D) TNF-α levels in plasma are unchanged by stress (n = 8, p = 0.59, left). Treatment with etanercept (Etan), a soluble blocking receptor for TNF-α, had no effect on stress-enhanced thrombus formation (n = 4, p = n.s., right). *p < 0.05.
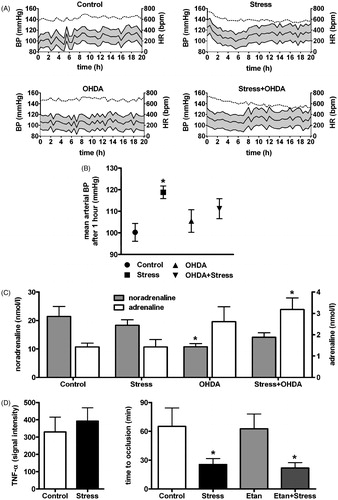
Noradrenaline (NA) levels were lower in plasma of 6-OHDA treated non-stressed mice as compared to vehicle treated non-stressed mice [n = 5, F(1,16) = 8.50, p < 0.05, ]. In stressed mice, the same treatment induced a tendency towards lower NA levels as well, but did not reach significance (n = 5, p = n.s., ). In both stressed and control mice, adrenaline (A) showed a trend towards higher levels in 6-OHDA treated mice as compared to vehicle treated mice, reaching significance in stressed mice [n = 5, F(1,26) = 10.07, p < 0.05, ]. Stress alone did not alter NA or A levels significantly (n = 5, p = n.s., ).
Plasma TNF-α levels remained unchanged after restraint stress as compared to controls (n = 8, p = 0.59, , left). Stressed mice treated with etanercept revealed a similar decrease in occlusion time as stressed animals treated with vehicle [n = 4, F(1,12) = 0.06, p = n.s., , right]. Immunohistochemical analysis revealed unaltered levels of the macrophage marker CD68 in carotid arteries of stressed mice as compared to controls [1.4 ± 0.4 versus 0.4 ± 0.16 stained cells/field, n = 5, F(1,16) = 1.75, p > 0.05, Supplementary Figure 1] irrespective of 6-OHDA treatment. Expression of the adhesion molecule VCAM-1 in carotid arteries of stressed mice was unchanged as compared to controls [6.6 ± 1.2 versus 3.2 ± 0.78 % of area, n = 5, F(1,16) = 1.63, p > 0.05, Supplementary Figure 1] irrespective of 6-OHDA treatment. There was no CD3, P-selectin or E-selectin detectable in any of the samples.
Discussion
The INTERHEART study analyzed the relationship between stress and myocardial infarction in >10,000 cases and is in line with other reports showing a clear correlation between stress and acute myocardial infarction (Rosengren et al., Citation2004). Restraint stress, a long established experimental model to assess stress related responses in mice (Yamamoto et al., Citation2002), was used in this study to investigate in vivo the effects of stress on arterial thrombus formation, the key event in acute coronary syndromes (Ross, Citation1999).
Stressed mice displayed a decreased time to thrombotic occlusion of the carotid artery, providing the first in vivo evidence for an increased prothrombotic potential as a direct response to stress.
To assess the role of the SNS, chemical sympathectomy was attained using 6-OHDA, which is known to induce degeneration of secondary sympathetic neurons. In vascular beds of rodents, destruction of adrenergic nerve endings has been shown to last for several weeks (Finch et al., Citation1972). 6-OHDA pretreatment (performed 2 days prior to the experiments) prevented the effect of stress on arterial thrombosis, indicating a pivotal role of the SNS in mediating the stress-induced increase in arterial thrombus formation.
TF, the main initiator of coagulation in vivo, is involved in the pathogenesis of arterial thrombus formation. Although it is predominantly known for its role in vascular smooth muscle cells, TF is also present in the circulating blood, referred to as blood-borne TF. Blood-borne TF is known to be biologically active (Bogdanov et al., Citation2003) and to originate from microparticles deriving from vascular smooth muscle cells, endothelial cells and leucocytes as well as from the alternatively spliced soluble form (Steffel et al., Citation2006). We observed elevated TF activity in plasma of stressed mice, suggesting that blood-borne TF promotes the enhanced thrombus formation after restraint stress. This increase in circulating TF was absent after chemical sympathectomy, indicating dependence on the SNS. In contrast to these findings, we observed that TF activity and expression in the vessel wall of stressed mice remained unchanged. This differs from another study demonstrating that TF mRNA was elevated in the adventitia of aortas from mice after restraint stress (Yamamoto et al., Citation2002). This discrepancy most likely results from different dissection techniques, since the adventitial tissue, which is known to express high loads of TF, was removed from the vessels in the present study. Furthermore, increased mRNA does not necessarily translate into an increased protein activity.
Platelets play a crucial role in thrombus formation and were previously shown to be affected by specific stressors (Larsson et al., Citation1989). In the present study platelet count, platelet aggregation to thrombin and tail bleeding time remained unaltered in stressed mice indicating that in this model, platelets are not a likely mediator of stress-enhanced arterial thrombosis. Plasma coagulation times (PT, representing the extrinsic, and aPTT, representing the intrinsic pathway of coagulation) as well as PAI-1 levels in plasma and t-PA expression in the vessel wall remained unchanged by restraint stress, indicating that plasmatic coagulation and fibrinolysis were not affected. In rats, PAI-1 was observed to increase within the first 3 hours of an immobilization stress protocol at the mRNA level (Ueyama et al., Citation2011). A transient increase in PA1-1 mRNA expression cannot be excluded in our mouse model, but did not translate into increased PAI-1 activity after 20 hours of restraint stress.
In view of the major role of the SNS in stress as well as in regulating arterial blood pressure, previous studies suggested that changes in blood pressure may mediate stress-induced adverse cardiovascular events (Dimsdale, Citation2008). Therefore, blood pressure and heart rate were measured telemetrically throughout the stress protocol. The areas under the curve for systolic, diastolic, and mean blood pressure as well as heart rate were comparable in all groups. Thus, we suggest that hemodynamic changes are unlikely to contribute to the enhanced thrombotic potential observed in stressed mice. The absence of relevant changes in blood pressure due to 6-OHDA treatment may seem surprising considering the blockade of the SNS under such conditions. However, similar findings were previously reported and explained by enhanced excretion of A by the adrenal medulla compensating for reduced NA levels, thereby restoring blood pressure to normal values (De Champlain & Van Ameringen, Citation1972). In line with these previous findings, NA was reduced after 6-OHDA treatment in the present study whilst A exhibited a trend towards higher plasma levels, reaching significance in stressed mice. The stress protocol itself had no influence on plasma catecholamine levels. In light of these observations, compensatory A excretion by the adrenal medulla may have prevented any major hemodynamic changes resulting from blockade of the SNS.
Systemic inflammation (as assessed by TNF-α measurement in plasma and TNF-α inhibition by a soluble blocking receptor) as well as local inflammation (as assessed by immunohistochemical analysis of CD68, VCAM-1, CD3, P-selectin and E-selectin expression in the arterial wall) revealed no changes in stressed mice (Supplemental Figure 1). In view of the synopsis of these experiments, we conclude that inflammation does not contribute the observed acceleration in thrombus formation.
Conclusion
We demonstrate that stress directly enhances arterial thrombosis in a controlled experimental setting. This effect is mediated through the SNS and at least in part by an increased activity of blood-born, but not vessel wall, TF. We cannot exclude that other parallel mechanisms contribute to the increased thrombotic response resulting from restraint stress and further studies will be needed to elucidate the molecular link between the SNS and the increase in plasma TF activity. Since high stress is a common feature characterizing modern societies, we are convinced that these novel findings may open future perspectives for the understanding of stress-induced cardiovascular events and thereby serve as a basis for clinical implications.
Declaration of interest
The study was supported by the Swiss National Science Foundation (to T.F.L.: 310030_118353, FCT: 310030_135781/1 and G.G.C.: 310030_130500), Velux Foundation, Wolfermann Nägeli Foundation, MERCATOR Foundation, Hartmann Müller Foundation, Olga Mayenfisch Foundation (to G.G.C.), Theodor und Ida Herzog-Egli Foundation and the Swiss Heart Foundation (to S.F.S.). The authors report no conflicts of interest.
Supplementary Material
Download PDF (229.6 KB)Related Research Data
References
- Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Nemerson Y. (2003). Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med 9(4):458–62
- Camici GG, Steffel J, Akhmedov A, Schafer N, Baldinger J, Schulz U, Shojaati K, et al. (2006). Dimethyl sulfoxide inhibits tissue factor expression, thrombus formation, and vascular smooth muscle cell activation: a potential treatment strategy for drug-eluting stents. Circulation 114(14):1512–21
- De Champlain J, Van Ameringen MR. (1972). Regulation of blood pressure by sympathetic nerve fibers and adrenal medulla in normotensive and hypertensive rats. Circ Res 31(4):617–28
- Dimsdale JE. (2008). Psychological stress and cardiovascular disease. J Am Coll Cardiol 51(13):1237–46
- Finch L, Haeusler G, Kuhn H, Thoenen H. (1972). Proceedings: the recovery of vascular adrenergic nerve function in the rat after chemical sympathectomy with 6-hydroxydopamine. Br J Pharmacol 44(2):357P–8P
- Glavin GB, Pare WP, Sandbak T, Bakke HK, Murison R. (1994). Restraint stress in biomedical research: an update. Neurosci Biobehav Rev 18(2):223–49
- Hui-Yuen JS, Duong TT, Yeung RS. (2006). Tnf-alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of kawasaki disease. J Immunol 176(10):6294–301
- Kohm AP, Sanders VM. (1999). Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo. J Immunol 162(9):5299–308
- Larsson PT, Hjemdahl P, Olsson G, Egberg N, Hornstra G. (1989). Altered platelet function during mental stress and adrenaline infusion in humans: evidence for an increased aggregability in vivo as measured by filtragometry. Clin Sci (Lond) 76(4):369–76
- Nawrot TS, Perez L, Kunzli N, Munters E, Nemery B. (2011). Public health importance of triggers of myocardial infarction: a comparative risk assessment. Lancet 377(9767):732–40
- Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. (2005). Defective thrombus formation in mice lacking coagulation factor xii. J Exp Med 202(2):271–81
- Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, Almahmeed WA, Blackett KN, et al. (2004). Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the interheart study): case-control study. Lancet 364(9438):953–62
- Ross R. (1999). Atherosclerosis – an inflammatory disease. N Engl J Med 340(2):115–26
- Stampfli SF, Akhmedov A, Gebhard C, Lohmann C, Holy EW, Rozenberg I, Spescha R, et al. (2010). Aging induces endothelial dysfunction while sparing arterial thrombosis. Arterioscler Thromb Vasc Biol 30(10):1960–7
- Steffel J, Luscher TF, Tanner FC. (2006). Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation 113(5):722–31
- Ueyama T, Yamamoto Y, Ueda K, Kawabe T, Hano T, Ito T, Tsuruo Y, et al. (2011). Cardiac and vascular gene profiles in an animal model of takotsubo cardiomyopathy. Heart Vessels 26(3):321–37
- Yamamoto K, Shimokawa T, Yi H, Isobe K, Kojima T, Loskutoff DJ, Saito H. (2002). Aging and obesity augment the stress-induced expression of tissue factor gene in the mouse. Blood 100(12):4011–18