
Abstract
Mechanisms of gastric defence under conditions of combined influence of acute stress and non-steroidal anti-inflammatory drugs (NSAIDs) are still poorly studied. The aim of this study was to explore the effects of different types of NSAIDs (naproxen, celecoxib and ATB-346) in producing experimental gastric lesions (induced by water-restraint stress (WRS) or by epinephrine (EPN) injection) and to determine the role of lipid peroxidation and the nitric oxide (NO) system in the pathogenesis of the damage. Male rats were used (eight per group) in this work. The NSAIDs were all administered at a dose 10 mg kg−1 30 min prior to WRS or EPN injection. Administration of naproxen to the control rats caused development of gastric lesions, whereas administration of a hydrogen sulfide (H2S)-releasing NSAID (ATB-346) or a selective cyclooxygenase-2 inhibitor (celecoxib) did not cause gastric damage. In contrast, lipid peroxidation processes were enhanced in all groups as was the activity of NO synthase (NOS). Pretreatment with naproxen in the WRS model caused an increase in severity of damage and a decrease in NOS activity. ATB-346 displayed beneficial effects, manifested by a decrease in the area of gastric damage, but parameters of lipid peroxidation and the NOS system did not differ substantially from those in the group treated with naproxen. Administration of different NSAIDs under conditions of EPN-induced gastric damage resulted in the decrease in NOS activity and lipid peroxidation. None of the tested NSAIDs exacerbated EPN-induced gastric mucosal injury; indeed, they all reduced the extent of damage.
Introduction
The human stomach is regularly exposed to a variety of noxious substances, including hydrochloric acid, refluxed bile salts, alcohol, ingested toxins or infectious agents such as Helicobacter pylori (Tarnawski et al., Citation2013). Several defence mechanisms are active in the gastric mucosa to protect it from injury, including the mucus-bicarbonate-phospholipid barrier, epithelial cells and gastric mucosal blood flow (Tulassay & Herszényi, Citation2010). The main components of mucosal defence occur at a molecular level, involving prostaglandins, polyamines, cytokines and gasotransmitters such as NO and H2S (Wallace, Citation2008). Nevertheless, when these protective mechanisms are overwhelmed by injurious factors, a gastric mucosal lesion may develop (Ham & Kaunitz, Citation2007).
Non-steroidal anti-inflammatory drugs (NSAIDs) can exert major detrimental effects on the gastric mucosa. In addition to inducing mucosal injury, these drugs can delay the healing of ulcers through a variety of mechanisms (Musumba et al., Citation2009). The most important of the systemic effects of NSAIDs is suppression of prostaglandin synthesis through inhibition of cyclo-oxygenase (COX) enzymes. COX-1 is the major source of prostaglandin synthesis in the stomach, but COX-2 also contributes significantly to prostaglandin-mediated mucosal defence (Wallace et al., 2000). Selective COX-2 inhibitors (coxibs) were developed based on the notion that COX-2 is solely responsible for the synthesis of prostaglandins that contribute to pain and inflammation, while COX-1 is mainly responsible for the synthesis of prostaglandins that mediate mucosal defence, but it is now clear that this was an oversimplification (Wallace et al., 2000). Furthermore, serious cardiovascular toxicity is associated with the use of selective COX-2 inhibitors and most conventional NSAIDs (naproxen being the exception) (Kearney et al., Citation2006). As a result, the search for safe NSAIDs has continued. In recent years, H2S-releasing NSAIDs are developed (Wallace & Vong, Citation2008; Wallace et al., Citation2010). H2S exerts strong anti-oxidative, anti-apoptotic, cytoprotective and anti-inflammatory activities in various tissues, including the gastric mucosa (Kimura et al., 2012; Predmore et al., Citation2012; Wallace et al., Citation2012). NSAIDs can reduce endogenous H2S synthesis (Fiorucci et al., Citation2005), while administration of H2S donors can protect the stomach against NSAID-induced gastric damage (Wallace et al., Citation2007).
Besides NSAIDs, another factor contributing to damage of the gastric mucosa is acute emotional stress. Therefore, stress models such as water-restraint stress (WRS) and injection of epinephrine (EPN) are widely used to induce gastric mucosal lesions in animals. These models seem to reproduce both local and systemic consequences of stress exposure to the upper gastrointestinal (GI) tract, resulting in the formation of bleeding gastric erosions and a decrease in mucosal blood flow. These effects could be caused by gastric vasoconstriction, leading to focal ischemia, hypoxia, and oxidative stress and thereby leading to formation of gastric lesions (Kwiecien et al., Citation2012). NSAID-induced adherence of leukocytes to the vascular endothelium has been shown to be an early and critical event in the pathogenesis of gastric injury, and may also contribute to the damage seen in stress-induced ulceration (Wallace et al., Citation1990).
The mechanisms involved in the pathogenesis of NSAID- and stress-induced ulcers are quite well studied. However, it is unknown what would happen to mechanisms of gastric defence and healing when the use of NSAIDs is accompanied by acute stress. For this reason, the purpose of our study was to explore the effects of different NSAIDs (conventional, selective COX-2 inhibitor and H2S-releasing) in experimental stress-related gastric lesions in rats, and to determine the role of lipid peroxidation and the nitric oxide (NO) system in ulcerogenesis and gastroprotection in rats. Thus, we assessed the effects of naproxen, celecoxib and ATB-346, representing the three types of NSAIDs, respectively. The results of our study support the hypothesis that ATB-346 may have beneficial effects in terms of GI safety as compared with other NSAIDs studied.
Methods
Animals
The structure of this study and the experimental procedures performed on the animals were approved by the Ethical Committee of Lviv National Medical University. The experimental procedures were carried out in accordance with international guidelines for the use and care of laboratory animals. Male, outbred wistar rats weighing 200–220 g were used. They were group-housed throughout (eight rats per cage) under conditions of controlled temperature (21–22 °C), humidity (65–75%) and light cycle (lights on at 08:00 h and off at 20:00 h) and fed standard rat chow and water ad libitum. Two models of gastric ulceration were used: water restraint stress (WRS) and EPN injection. The rats were fasted for 24 h prior to the experimental procedures.
WRS-induced gastric mucosal lesions
Rats were restrained in wire cages and immersed up to the depth of the xiphoid process in a water bath (23 °C) for 5 h to induce gastric mucosal lesions, as described by Takagi et al. (Citation1964).
EPN-induced gastric damage
EPN was administered intraperitoneally at a dose of 2 mg/kg (injectate concentration 1.8 mg/mL), 24 h later the rats were killed (Zhuroms’kyĭ & Sklyarov, Citation2011).
Test drugs
Naproxen, ATB-346 and celecoxib were examined in both models of gastric damage. Before administration, the drugs were dissolved in a small amount of DMSO, then suspended in 1% carboxymetylcellulose. Naproxen, ATB-346 and celecoxib were administered intragastrically (via an orally introduced polyethylene tube) at a single dose (10 mg kg−1; volume of 1 mL) 30 min prior to WRS or EPN injection. These procedures were initiated between 09:00 and 09:30 h. This dose was selected because it produced significant and comparable anti-inflammatory activity in reducing swelling in experimental arthritis (Blackler et al., Citation2012).
Study protocol
The rats were randomly divided into 12 groups (n = 8 in each group). Group 1 consisted of rats treated only with vehicle. Other groups of rats were pretreated with vehicle (groups 5 and 9), naproxen (Sigma-Aldrich, Milwaukee, WI; groups 2, 6 and 10), ATB-346 (Antibe Therapeutics Inc., Toronto, Canada; groups 3, 7 and 11), or celecoxib (Antibe Therapeutics Inc., Toronto, Canada; groups 4, 8 and 12). The 2nd–4th groups were used to study the influence of drugs alone. The 5th–8th groups were subjected to WRS for 30 min after treatment with the above-mentioned drugs. The 9th–12th groups were administered EPN 30 min after treatment with the above-mentioned drugs.
Rats were anesthetized with 1 mL of urethane at a dose of 1.1 g/kg injected intraperitoneally and killed by cervical dislocation. Urethane was used because it does not have the suppressive effect on gastric acid secretion that is seen with other commonly used anaesthetics. A trunk blood sample after decapitation was immediately collected into vials containing 0.1 mL of heparin. The stomach was then harvested, opened along the lesser curvature, and washed in isotonic sodium chloride solution. The severity of mucosal lesions was macroscopically scored by an observer blind to the treatments the rats had received. The area (in mm2) of lesions was measured by planimetry and the severity of damage scored according to the following criteria: (1) hyperemia: score 2; (2) petechiae (small hemorrhages): score 4; (3) hemorrhages and superficial erosion: score 6; (4) several superficial erosions or one deep erosion: score 8; (5) deep, bleeding lesions: score 10; and (6) deep, bleeding lesions, erosions and hemorrhages: score 12 (Nasadyuk & Sklyarov, Citation2013). Gastric mucosal samples from the whole mucosa were collected and homogenized in saline (1:4), then centrifuged at 2000g for 15 min at 4 °C; the supernatant was used for the measurement of various biochemical parameters (see below).
Biochemical assessment
Lipid peroxidation levels were determined as malonic dialdehyde (MDA) concentration in homogenates of gastric mucosa, according to the procedure of Timirbulatov & Seleznev (Citation1981), which measures concentrations of MDA in the range of 50–500 μmol/g. The concentration of NO in gastric mucosa was assessed as the content of nitrite anion using the Griess assay (Green et al., 1982) with a sensitivity for the measurement of nitrite anions concentrations over 5 μmol/g of tissue. NO synthase (NOS) activity (total NOS, inducible NOS (iNOS), and constitutive NOS (cNOS)) was measured by the method described in detail previously (Sklyarov et al., Citation2011) and was expressed in nmol NADPH/min g of protein. Arginase activity was determined by the method Geyer & Dabich (Citation1971) and was expressed in μmol/min mg of protein. The concentration of l-arginine in plasma samples was measured according to the method of Alejnikova et al. (Citation1988) and was sensitive to measure concentrations of l-arginine in the range of 5–50 μg/mL.
Statistical analyses
The data are expressed as mean ± SD. Comparisons involving more than two groups were performed by a one-way analysis of variance (ANOVA), Fisher’s Protected Least Significant Difference test was used (degrees of freedom, F and p values were determined). Differences with p value <0.05 were considered as significant.
Results
Effect of NSAIDs on the macroscopic mucosal damage and parameters of lipoperoxidation and NO-system
Visual inspection showed that there were no gastric mucosal lesions in vehicle-treated rats (group 1), whereas essentially all rats treated with naproxen developed ulcerative gastric lesions and erosions (F(4.41) = 10.1, p < 0.01, and ). In contrast, ATB-346 did not induce significant gastric damage, consistent with previously published data, where H2S was shown to act as a mediator in mucosal defence and administration of H2S-releasing NSAIDs produces substantially less GI injury than conventional NSAIDs (Wallace et al., Citation2010).
Figure 1. Representative photographs of the gastric mucosa of drug alone groups: (A) vehicle; (B) naproxen; (C) ATB-346; (D) celecoxib. Arrow indicates the location of gastric damage.
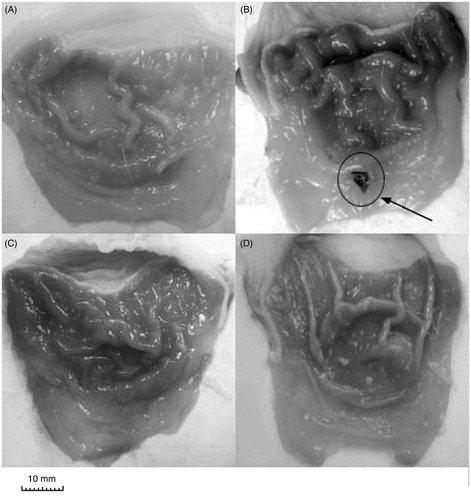
Figure 2. Area (panel A) and score (panel B) of the gastric lesions of rats treated with vehicle, naproxen, ATB-346 or celecoxib. Results are expressed as the mean ± SD for eight rats per group; *p < 0.05, **p < 0.01 versus the indices of vehicle-treated group (data were compared using ANOVA Fisher’s Protected Least Significant Difference test, differences with p value <0.05 were considered as significant).
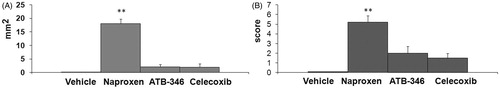
Celecoxib did not cause significant changes in gastric mucosal integrity, corresponding with the data reported previously that selective COX-2 inhibitors produce severe GI complications less frequently than conventional NSAIDs, but the gastric mucosa of this group of rats was obscured by an insoluble white mucous layer. We consider that the appearance of this mucus could be caused by COX-2 inhibition and consequently by up-regulation of growth factor regulating the regeneration of the mucosa (Musumba et al., Citation2009) and/or impairment of gastric mucus production (Wallace et al., Citation2011).
There was significant activity of cNOS in gastric mucosa of vehicle-treated rats while iNOS activity was low (), consistent with the data reported by other authors (Nasadyuk & Sklyarov, Citation2013). Naproxen increased total NOS activity in gastric mucosa (F(4.96) = 5.73, p = 0.02), mainly due to the 3.5-fold increase in iNOS activity (F(4.96) = 19.59, p < 0.01). cNOS activity practically did not change. Concomitantly, activity of arginase in gastric mucosa decreased, but this change of activity was not statistically significant as also were changes in nitrite anion concentration and l-arginine level in blood. ATB-346 caused similar effects to its parent drug, naproxen, on parameters of NOS activity: a tendency to increased total NOS and a statistically significant increase in iNOS (F(4.96) = 11.07, p = 0.001) activities. The selective COX-2 inhibitor, celecoxib, also elicited an increase in iNOS activity (p < 0.01 F(4.96) = 10.55). The concentration of a final product of lipid peroxidation, MDA, was significantly (p < 0.01) increased in all groups (F(4.96) = 12.2 for naproxen, F(4.96) = 11.06 for ATB-346 and F(4.96) = 9.9 for celecoxib) treated with the different NSAIDs, indicating the intensification of oxidative processes.
Table 1. Effect of NSAIDs (conventional – naproxen; selective cyclooxygenase-2 inhibitor – celecoxib; and hydrogen sulphide-releasing – ATB-346) on concentration of MDA, nitrite anion, activity of NOS and arginase in gastric mucosa and concentration of l-arginine in blood.
WRS-induced gastric mucosal damage
Gross analysis showed that 5 h of WRS induced the development of severe bleeding erosions in the stomach ().
Figure 3. Representative photographs of the gastric mucosa of drug + stress groups: (A) vehicle + water-restraint stress (WRS), (B) naproxen + WRS, (C) ATB-346 + WRS, (D) celecoxib + WRS. Arrows indicate the main areas of gastric damage.
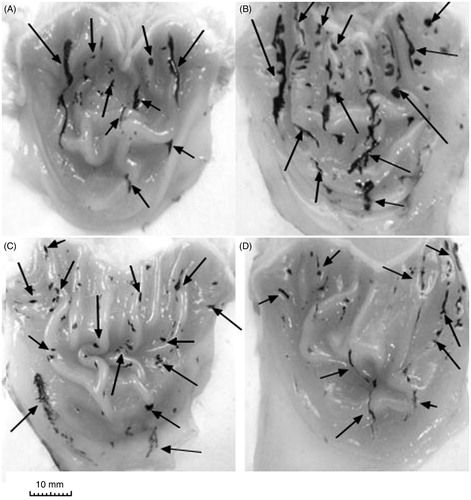
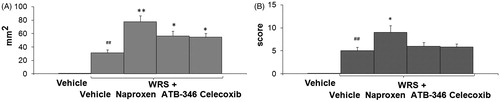
Pretreatment with naproxen potentiated the development of gastric lesions caused by WRS with a significant increase in the gastric lesion area (2.5-fold, F(4.41) = 16.9, p < 0.01) (). The area of gastric lesions induced by WRS in rats pretreated with ATB-346 was significantly lower than that in the group pretreated with the parent NSAID, naproxen (). However, that damage was still much higher than in the vehicle + WRS group (F(4.41) = 6.5, p = 0.034). Similarly, rats pretreated with the selective COX-2 inhibitor celecoxib prior to WRS developed gastric damage that was more extensive than that in the group treated with vehicle prior to WRS (F(4.41) = 5.11, p < 0.045).
Figure 4. Area (panel A) and score (panel B) of the gastric lesions of drug + stress groups: vehicle-treated; vehicle + WRS; naproxen + WRS; ATB-346 + WRS; celecoxib + WRS. Results are expressed as the mean ± SD for eight rats per group; *p < 0.05, **p < 0.01 versus the indices in vehicle + WRS; #p < 0.05, ##p < 0.01 versus the vehicle-treated rats (data were compared using ANOVA Fisher’s Protected Least Significant Difference test, differences with p value <0.05 were considered as significant).
The gastric mucosa of rats treated with vehicle then subjected to WRS was characterized by the following changes in biochemical indices: enhanced activity of lipid peroxidation processes manifested by increases in MDA (F(4.41) = 10.1, p < 0.01) and nitrite anion (F(4.41) = 9.6, p < 0.01) concentrations (). Administration of naproxen, ATB-346 or celecoxib did not significantly change these parameters.
Table 2. Effect of NSAIDs (conventional – naproxen; selective cyclooxygenase-2 inhibitor – celecoxib; and hydrogen sulphide-releasing – ATB-346) under WRS on concentration of MDA, nitrite anion, activity of NOS and arginase in gastric mucosa and concentration of l-arginine in blood.
Subjecting rats to WRS resulted in a considerable increase in NO synthase activity (F(4.41) = 12.11, p < 0.01), especially iNOS (F(4.41) = 8.01, p = 0.0002), and a concomitant decrease in arginase activity (F(4.41) = 10.06, p < 0.01). Likely a consequence of the increased activation of NO synthases, concentrations of l-arginine, the substrate for NOS, decreased in the plasma (F(4.41) = 7.75, p = 0.012). Administration of naproxen prior to WRS resulted in a significant decrease in the total NOS activity (F(4.67) = 12.09, p < 0.01), caused by statistically significant decreases in both isoforms of this enzyme (iNOS [F(4.67) = 19.01, p = 0.0002] and cNOS [F(4.67) = 5.11, p = 0.04]) in the stomach as compared with the vehicle + WRS group (). The activity of arginase remained lower than normal. Indices of the NO synthase system in ATB-346-pretreated rats subjected to WRS were similar to those of the group treated with naproxen + WRS. Celecoxib + WRS did not affect the parameters of the NO synthase system as compared with WRS alone; only nitric anion concentration increased significantly (F(4.96) = 6.07, p = 0.021).
Model of EPN-induced gastric damage
Visual inspection showed that all rats subjected to EPN injection developed multiple ulcerative and hemorrhagic gastric lesions (). Neither naproxen nor celecoxib caused significant differences in gastric damage compared with the EPN only group (). Only pre-treatment with ATB-346 led to attenuation of the EPN-induced damage (both the damage area and the damage score decreased: F(4.41) = 5.12, p = 0.031 for damage area).
Figure 5. Representative photographs of the gastric mucosa of drug + EPN groups: (A) vehicle + EPN; (B) naproxen + EPN; (C) ATB-346 + EPN; (D) celecoxib + EPN. Arrows indicate the main areas of gastric damage.
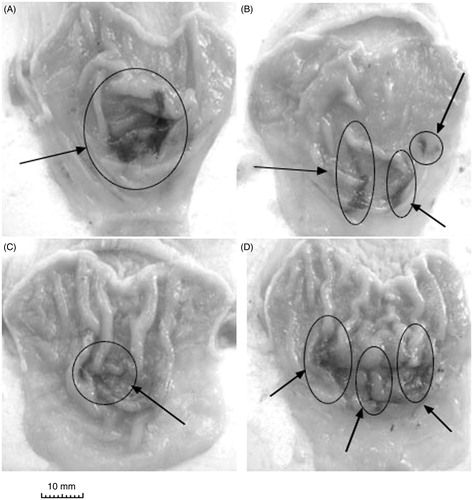
Figure 6. Area (panel A) and score (panel B) of the gastric lesions of drug + EPN groups: vehicle-treated; vehicle + EPN; naproxen + EPN; ATB-346 + EPN; celecoxib + EPN. Results are expressed as the mean ± SD for eight rats per group; *p < 0.05 versus the indices in vehicle + EPN; ##p < 0.01 versus the vehicle group (data were compared using ANOVA Fisher’s Protected Least Significant Difference test, differences with p value <0.05 were considered as significant).
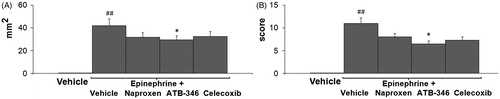
The rats exposed to the ulcerogenic effect of EPN showed an increase in MDA concentration in gastric mucosa (F(4.41) = 11.9, p < 0.01), indicating oxidative stress caused by ischemia and hypoxia. Under the same conditions the concentration of nitrite anion also increased (). All tested NSAIDs (naproxen, ATB-346 and celecoxib) decreased the level of MDA by approximately the same amounts (F(4.41) = 14.15, p < 0.01 for naproxen, F(4.41) = 9.4, p < 0.01 for ATB-346 and F(4.41) = 16.64, p < 0.01 for celecoxib) as well as nitrite anion concentration (F(4.41) = 16.15, p < 0.01 for naproxen, F(4.41) = 12.1, p < 0.01 for ATB-346 and F(4.41) = 11.49, p < 0.01 for celecoxib).
Table 3. Effect of NSAIDs (conventional – naproxen; selective cyclooxygenase-2 inhibitor – celecoxib; and hydrogen sulphide-releasing – ATB-346) under EPN-induced gastric mucosal damage on concentration of MDA, nitrite anion, activity of NOS and arginase in gastric mucosa and concentration of l-arginine in blood.
The development of the EPN-induced gastric lesions in rats was accompanied by a sharp increase in the total NOS activity in gastric mucosa, which increased 3.6-fold (F(4.41) = 10.8, p < 0.01), mainly due to the 10-fold increase in iNOS activity (F(4.41) = 13.4, p < 0.01), whereas cNOS activity showed only a tendency to increase as compared with the control group. Concomitantly, both l-arginine concentration in blood plasma and activity of arginase in gastric mucosa decreased (F(4.41) = 13.9, p = 0.0025, for l-arginine and F(4.41) = 4.99, p = 0.04 for arginase) (). Administration of naproxen prior to EPN reduced indices of the NO synthase system in gastric mucosa as compared to action of EPN alone: NOS activity was lower mainly due to reduced iNOS activity (F(4.41) = 10.86, p < 0.01). The l-arginine level in blood was essentially normal. A similar tendency toward changes in NOS activity parameters was noted in the ATB-346 + EPN group. However, arginase activity was significantly increased (F(4.41) = 5.11, p = 0.033) as compared with the vehicle + EPN group.
Celecoxib administration prior to EPN injection caused a decrease in NOS activity (F(4.41) = 13.02, p < 0.01) as compared with the vehicle + EPN group. Gastric arginase activity increased as compared with the vehicle + EPN group (F(4.41) = 7.17, p = 0.012).
Discussion
In this study, we have examined the action of different NSAIDs, conventional (naproxen), H2S-releasing (ATB-346) and selective COX-2 inhibiting (celecoxib), in healthy rats and rats with experimental stress-related gastric lesions and following EPN injection. Our experiments demonstrated a clear beneficial effect of ATB-346 over the other drugs with respect to the extent of gastric damage that was elicited by their administration.
Gastric ulcer disease remains widespread: NSAIDs and stressful lifestyle make significant contributions to this pathological situation (Filaretova et al., Citation2007). NSAIDs can impair mucosal defence by suppressing prostaglandin synthesis (Takeuchi, Citation2012). This renders the stomach more susceptible to injury (Wallace & Vong, Citation2008). In the present study, the administration of naproxen at a dose of 10 mg kg−1 caused the development of ulcerative lesions and erosions in the gastric mucosa. This ulcerogenic action of naproxen is a result of combination of its topical and systemic effects, described previously (Musumba et al., Citation2009; Wallace, Citation2008). In contrast, a novel H2S-releasing derivative of naproxen, ATB-346 [2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester] did not induce significant gastric damage. It was previously shown that H2S serves as a mediator in mucosal defence and administration of H2S-releasing NSAIDs produces substantially less GI injury than conventional NSAIDs (Wallace et al., Citation2007, Citation2010).
In our research, the selective COX-2 inhibitor celecoxib did not cause serious changes in gastric mucosal integrity, corresponding with the data reported previously that selective COX-2 inhibitors produce severe GI complications less frequently than conventional NSAIDs. But it should be pointed out that the gastric mucosa of rats treated with celecoxib was overlaid by an insoluble white mucous layer. This mucus may have resulted in an up-regulation of growth factor release (Musumba et al., Citation2009) and activation of regeneration processes and/or impairment of gastric mucus production (Wallace et al., Citation2011).
The generation of free radicals may be a major factor underlying peptic ulceration, including the ulceration induced by NSAIDs. There is substantial evidence that oxygen-derived free radicals play an important role in the pathogenesis of gastric mucosal injury (Yoshikawa et al., Citation1993). In addition, involvement of oxygen-derived free radicals such as superoxide anion, hydrogen peroxide and hydroxyl radical are well established in the pathogenesis of ischemic injury of GI mucosa (Kwiecien et al., Citation2012). In this study, the concentration of a final product of lipid peroxidation, MDA, was increased in the gastric mucosa of all groups treated with the different NSAIDs, indicating an intensification of oxidative processes. It was previously reported that H2S inhibits oxidative damage of tissue, in part through scavenging of peroxinitrite and other oxygen-derived free radicals (Liu et al., Citation2012). However, in our study ATB-346 at the dose of 10 mg kg−1 did not decrease MDA. Thus, H2S released from this NSAID may have acted through other mechanisms of cytoprotection, such as suppression of leukocyte adherence to the vascular endothelium and reduction of leukocyte extravasation and oedema formation, shown previously (Wallace, Citation2008).
Another important system involved in regulation of both cytoprotection and ulcerogenesis is the NO system (Lunberg & Weitzberg, Citation2013). It is widely accepted that constitutive forms of NOS, neuronal NOS (nNOS) and endothelial NOS (eNOS), are important in the normal function of the GI tract in that inhibition of these enzymes can result in disturbances of GI motility, blood flow and secretion. However, iNOS, which produces relatively large amounts of NO under certain pathological conditions, contributes to mucosal injury and dysfunction (Sklyarov et al., Citation2011). The toxicity of NO has been attributed to the potent nitrating and oxidizing agent, peroxynitrite, which is formed by a reaction between NO and superoxide (Ohta & Nishida, Citation2002). In the present study in the control group of rats, the activity of cNOS in gastric mucosa was prevalent and iNOS activity was low, corresponding with the data reported by other authors (Nasadyuk & Sklyarov, Citation2013). Oral administration of naproxen at the dose of 10 mg kg−1 increased the activity of NO synthases. Due to activation of NO synthases, the concentrations of l-arginine in the blood plasma decreased and content of nitrite anion in the gastric mucosa increased. Both naproxen and ATB-346 increased the activity of NOS (total and iNOS) to the same extent. These changes in the NO system parameters can be explained by the close inter-relationship existing between iNOS and COX in the process of cytoprotection of the gastric mucosa (Mollace et al., Citation2005). Such acute increases in enzymatic activity of iNOS and increased generation of nitrite anion were previously shown in indomethacin-induced models of gastric ulceration (Nasadyuk & Sklyarov, Citation2013). In the present study, we showed that H2S released from ATB-346 probably did not play the pivotal role in activation of iNOS, but it was evidently involved in other mechanisms (NOS-independent) of gastroprotection.
WRS produces a combination of different events. On the one hand, it triggers the release of catecholamines (EPN and norepinephrine), which lead to a decrease in submucosal and mucosal blood flow, local ischemia and impairment of tissue resistance, resulting in gastric lesions (Kwiecien et al., Citation2012). On the other hand, glucocorticoid production also increases under conditions of stress (Filaretova et al., Citation2012). These hormones can inhibit phospholipase A2, an enzyme responsible for the release of arachidonic acid, which serves as a substrate for the COX enzymes. Thus, WRS can induce a significant inhibition of PGE generation by the gastric mucosa (Basso et al., Citation1983). It was recently shown that glucocorticoids can exert gastroprotective actions associated with maintenance of general body homeostasis, including blood glucose levels and systemic blood pressure (Filaretova et al., Citation2007). The destructive changes that the gastric mucosa is subjected to due to WRS were associated with increased numbers of active forms of oxygen, enhanced synthesis of NO, increased expression of iNOS by epithelial cells and macrophages, and neutrophil infiltration into the damaged mucous membrane. On the one hand, pretreatment with naproxen in the WRS model in rats caused an increase in the area damaged and in severity of damage. On the other hand, there was a decrease in NOS activity (both isoforms). This inhibition of NO synthesis could impair gastric mucosal defence resulting in tissue injury. These effects of naproxen can be explained by its topical and systemic effects, connected with the inhibition of COX (Musumba et al., Citation2009) as well as with the hypoxia of epithelial cells, caused by vasoconstriction. We suggest that the action of naproxen under conditions of overproduction of glucocorticoids in stress sharply reduces the synthesis of prostaglandins, playing a crucial role in damage to the gastric mucosa. This hypothesis corresponds with the data reported previously that hypoglucocorticism caused by adrenalectomy markedly worsens the gastric erosion induced by NSAIDs (Filaretova, Citation2013).
ATB-346 displayed a beneficial effect, manifested by a decrease in total area of gastric damage. However, the parameters of lipoperoxidation and NO synthase activity did not differ substantially from those in the group treated with naproxen prior to WRS. We hypothesize that the reduction of gastric damage with an H2S-releasing NSAID is a result of its maintenance of gastric blood flow and inhibition of leukocyte adherence (Zanardo et al., Citation2006). It should also be pointed out that there is an interrelationship between H2S and NO in the regulation of biological functions. It has been shown that H2S exerts anti-inflammatory protection of organs through an eNOS/NO and p38 mitogen-activated protein kinase-dependent mechanism (Kolluru et al., Citation2013). However, in our study, ATB-346 being administered only as a single dose may have been insufficient to observe any Н2S-related effects on the NOS system.
It was previously shown that pretreatment with a COX-2 inhibitor (rofecoxib) aggravated gastric damage caused by 3.5 h of WRS, but did not significantly change MDA levels (Kwiecien et al., Citation2012). However, in our study, inhibition of COX-2 prior to WRS was associated with a significant exacerbation of gastric damage, though less severe than observed in rats in the naproxen + WRS group. Celecoxib caused a considerable increase in NOS activity as compared with rats treated with WRS alone.
Although stress is a complex state, it is clear that EPN is a significant contributor to stress-related GI damage. It is well known that an acute rise of the EPN concentration in blood causes the development of hypoxia in the stomach, the release of lysosomal enzymes and an increase in the level of reactive oxygen species (Nasadyuk & Sklyarov, Citation2013). In the present study, injection of EPN resulted in the development of abundant hemorrhagic lesions of the gastric mucosa, an acute rise in iNOS activity, increased generation of NO, decreased activity of arginase and the development of oxidative damage in the gastric mucosa. Administration of different NSAIDs under conditions of EPN-induced “stress” resulted in similar effects, manifested by a decrease in area and the score for the gastric damage, as well as activity of NOS and intensity of lipid peroxidation. In contrast to the WRS model, none of the tested NSAIDs administered prior to EPN injection exacerbated gastric mucosal injury; indeed, they all reduced the extent of damage.
Conclusions
Both the WRS and EPN models of gastric damage in rats were accompanied by the activation of iNOS activity and the development of oxidative stress manifested by an increase in MDA concentrations. Pretreatment with a conventional NSAID, naproxen, exacerbated gastric lesions in the WRS model and reduced them in the EPN model. It also decreased iNOS activity in both models. The gastric lesions in rats pretreated with an H2S-releasing NSAID (ATB-346) in both models showed beneficial effects, because the areas and scores of gastric damage were less than those observed in rats treated with the COX-2-selective inhibitors, celecoxib, regarded as the safest in terms of GI damage of currently marketed NSAIDs. However, parameters of lipid peroxidation and the NO synthase system did not differ substantially from those in the group treated with naproxen prior to WRS, proving the dominant role of the parent drug influence on these indices.
Declaration of interest
Other than Dr Wallace, who is a founder and director of Antibe Therapeutics Inc., the authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Alejnikova TL, Rubtsova GV, Pavlova NA. (1988). Manuals for practical lessons in biochemistry. Moscow: Medicine
- Basso N, Materia A, Forlini A, Jaffe BM. (1983). Prostaglandin generation in the gastric mucosa of rats with stress ulcer. Surgery 94(1):104–8
- Blackler R, Syer S, Bolla M, Ongini E, Wallace JL. (2012). Gastrointestinal-sparing effect of novel NSAIDs in rats with compromised mucosal defence. PLoS One 7:e35196
- Filaretova L, Bagaeva T, Morozova O. (2012). Stress and the stomach: corticotropin-releasing factor may protect the gastric mucosa in stress through involvement of glucocorticoids. J Physiol Pharmacol 63:143–53
- Filaretova L, Podvigina T, Bagaeva T, Bobryshev P, Takeuchi K. (2007). Gastroprotective role of glucocorticoid hormones. J Pharmacol Sci 104:195–201
- Filaretova L. (2013). Gastroprotective role of glucocorticoids during NSAID-induced gastropathy. Curr Pharm Des 19:29–33
- Fiorucci S, Antonelli E, Distrutti E, Mencarelli A, Orlandi S, Zanardo R, Renga B, et al. (2005). Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 129:1210–24
- Geyer JW, Dabich D. (1971). Rapid method for determination of arginase activity in tissue homogenates. Anal Biochem 39:412–7
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. (1982). Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–8
- Ham M, Kaunitz JD. (2007). Gastroduodenal defense. Curr Opin Gastroenterol 24:607–16
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. (2006). Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomized trials. BMJ 332:1302–08
- Kimura H, Shibuya N, Kimura Y. (2012). Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal 17:45–57
- Kolluru GK, Shen X, Kevil G. (2013). A tale of two gases: NO and H2S, foes or friends for life? Redox Biol 1:313–8
- Kwiecien S, Konturek PC, Sliwowski Z, Mitis-Musiol M, Pawlik MW, Brzozowski B, Jasnos K, et al. (2012). Interaction between selective cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress. J Physiol Pharmacol 63:143–51
- Liu L, Cui J, Song ChJ, Bian JS, Sparatore A, Soldato PD, Wang XYu, et al. (2012). H2S-releasing aspirin protects against aspirin-induced gastric injury via reducing oxidative stress. PLoS One 7:e46301
- Lunberg JО, Weitzberg E. (2013). Biology of nitrogen oxides in the gastrointestinal tract. Gut 62:616–26
- Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. (2005). Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev 57:217–52
- Musumba C, Pritchard DM, Pirmohamed M. (2009). Review article: cellular and molecular mechanisms of NSAID-induced peptic ulcers. Aliment Pharmacol Ther 30:517–31
- Nasadyuk C, Sklyarov AY. (2013). Thymohexin exhibits cytoprotective effect in experimental gastric lesions in rats both through the inhibition of inducible nitric oxide synthase and reduction of oxidative mucosal damage. Regul Pept 180:50–7
- Ohta Y, Nishida K. (2002). l-Arginine protects against stress-induced gastric mucosal lesions by preserving gastric mucus. Clin Exp Pharmacol Physiol 29:32–8
- Predmore BL, Lefer DJ, Gojon G. (2012). Hydrogen sulfide in biochemistry and medicine. Antioxid Redox Signal 17:119–40
- Sklyarov AY, Panasyuk NB, Fomenko IS. (2011). Role of nitric oxide-synthase and cyclooxygenase/lipooxygenase systems in development of experimental ulcerative colitis. J Physiol Pharmacol 62:65–73
- Takagi K, Kasuya Y, Watanabe K. (1964). Studies on the drugs for peptic ulcer. A reliable method for producing stress ulcer in rats. Chem Pharm Bull 12:465–72
- Takeuchi K. (2012). Pathogenesis of NSAID-induced gastric damage: importance of cyclooxygenase inhibition and gastric hypermotility. World J Gastroenterol 18:2147–60
- Tarnawski A, Ahluwalia A, Jones MK. (2013). Gastric cytoprotection beyond prostaglandins: cellular and molecular mechanisms of gastroprotective and ulcer healing actions of antacids. Curr Pharm Des 19:126–32
- Timirbulatov RA, Seleznev EI. (1981). Method for increasing the intensity of free radical oxidation of lipid-containing components of the blood and its diagnostic significance. Lab Delo 4:209–11
- Tulassay Z, Herszényi L. (2010). Gastric mucosal defense and cytoprotection. Best Pract Res Clin Gastroenterol 24:99–108
- Wallace JL. (2008). Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev 88:1547–65
- Wallace JL, Caliendo G, Santagada V, Cirino G. (2010). Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol 159:1236–46
- Wallace JL, Dicay M, McKnight W, Martin GM. (2007). Hydrogen sulfide enhances ulcer healing in rats. FASEB J 21:4070–6
- Wallace JL, Ferraz JG, Muscara MN. (2012). Hydrogen sulfide: an endogenous mediator of resolution of inflammation and injury. Antioxid Redox Signal 17:58–67
- Wallace JL, Keenan CM, Granger DN. (1990). Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol 1259:G462–7
- Wallace JL, McKnight W, Reuter BK, Vergnolle N. (2000). NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 119(3):706–14
- Wallace JL, Vong L. (2008). NSAID-induced gastrointestinal damage and the design of GI-sparing NSAIDs. Curr Opin Investig Drugs 9:1151–6
- Wallace JL, Vong L, Dharmani P, Srivastava V, Chadee K. (2011). Muc-2-deficient mice display a sex-specific, COX-2-related impairment of gastric mucosal repair. Am J Pathol 178:1126–33
- Yoshikawa T, Naito Y, Nakamura S, Nishimura S, Kaneko T, Iinuma S, Takahashi S, et al. (1993). Effect of rebamipide on lipid peroxidation and gastric mucosal injury induced by indometacin in rats. Arzneimittelforschung 43:1327–30
- Zanardo RCO, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. (2006). Hydrogen sulphide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J 20:2118–20
- Zhuroms’kyĭ VS, Sklyarov AY. (2011). Effect of vitamin C on the condition of NO-synthase system in experimental stomach ulcer. Fiziol Zh 57:90–8