
Abstract
We hypothesized that acute stress would induce endothelial dysfunction. Male Wistar rats were restrained for 2 h within wire mesh. Functional and biochemical analyses were conducted 24 h after the 2-h period of restraint. Stressed rats showed decreased exploration on the open arms of an elevated-plus maze (EPM) and increased plasma corticosterone concentration. Acute restraint stress did not alter systolic blood pressure, whereas it increased the in vitro contractile response to phenylephrine and serotonin in endothelium-intact rat aortas. NG-nitro-l-arginine methyl ester (l-NAME; nitric oxide synthase, NOS, inhibitor) did not alter the contraction induced by phenylephrine in aortic rings from stressed rats. Tiron, indomethacin and SQ29548 reversed the increase in the contractile response to phenylephrine induced by restraint stress. Increased systemic and vascular oxidative stress was evident in stressed rats. Restraint stress decreased plasma and vascular nitrate/nitrite (NOx) concentration and increased aortic expression of inducible (i) NOS, but not endothelial (e) NOS. Reduced expression of cyclooxygenase (COX)-1, but not COX-2, was observed in aortas from stressed rats. Restraint stress increased thromboxane (TX)B2 (stable TXA2 metabolite) concentration but did not affect prostaglandin (PG)F2α concentration in the aorta. Restraint reduced superoxide dismutase (SOD) activity, whereas concentrations of hydrogen peroxide (H2O2) and reduced glutathione (GSH) were not affected. The major new finding of our study is that restraint stress increases vascular contraction by an endothelium-dependent mechanism that involves increased oxidative stress and the generation of COX-derived vasoconstrictor prostanoids. Such stress-induced endothelial dysfunction could predispose to the development of cardiovascular diseases.
Introduction
Psychosocial stress is associated with increased cardiovascular morbidity and mortality (Bosma et al., Citation1997). Chronic stress induces hypertension and it is proposed that endothelial dysfunction is a key factor in the development of this response (Chung et al., Citation2010; Esler et al., Citation2008). In line with this, chronic stress decreases the activity of nitric oxide (NO) synthase and the vascular generation of NO with consequent increase in vasocontractile responses (Neves et al., Citation2009; Okruhlicová et al., Citation2008). Moreover, chronic stress-induced hypertension and endothelial dysfunction are associated with nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidase activation and oxidative stress (Chung et al., Citation2010).
Acute stress is also described to increase blood pressure (Crestani et al., Citation2010; McDougall et al., Citation2005; Tavares & Correa, Citation2006), but little is known about its effects in the vasculature. The vascular endothelium is important in the maintenance of vascular tone as it produces endothelial-derived mediators, which are involved in the contraction and relaxation of the vasculature (Tousoulis et al., Citation2014). Endothelial dysfunction results in impaired endothelium-mediated vasodilatation and increased vascular reactivity, and is associated with several pathologies in the cardiovascular system. Increased generation of prostanoids and reactive oxygen species (ROS) together with decreased NO bioavailability are important molecular events underlying endothelial dysfunction (Tousoulis et al., Citation2014). Interestingly, acute stress was described to reduce NO generation by alveolar macrophages (Persoons et al., Citation1995). Additionally, acute stress was also shown to reduce plasma NO level in humans, increase plasma lipid peroxidation and reduce endogenous antioxidant systems (Liu & Mori, Citation1994; Liu et al., Citation1994; Matsumoto et al., Citation1999; Yeh et al., Citation2002). Finally, acute stress was described to increase ROS generation and lipid peroxidation in the gastric and intestinal mucosa of rats (Bagchi et al., Citation1999). With regard to vascular effects, acute stress was shown to induce a corticosterone-dependent increase in prostacyclin (PGI2) concentration in the rat aorta (Grimée & Wülfert, Citation1995) and to reduce vascular relaxation in healthy subjects without overt vascular disease (Ghiadoni et al., Citation2000). Increased expression of vascular-endothelial growth factor (VEGF) was also described after acute stress (Manni et al., Citation2005). However, whether acute stress increases ROS generation and reduces NO bioavailability in the vasculature remains elusive.
Several stressful stimuli can be used to induce cardiovascular responses. Restraint has been standardized in the literature as an unavoidable aversive stimulus eliciting sustained blood pressure increase in rats (Crestani et al., Citation2010; McDougall et al., Citation2005). Moreover, restraint stress is associated with well-characterized behavioral changes, including reduced exploratory activity in the elevated plus-maze (EPM) (McBlane & Handley, Citation1994; Padovan & Guimarães, Citation2000). Although restraint-induced stress increases in blood pressure are well documented, little information about vascular changes associated with this model of psychological stress is available. Since acute stress reduces tissue and plasma NO bioavailability and increases ROS generation, we hypothesized that acute stress would induce endothelial dysfunction. In the present study, we investigated the effect of acute stress in the responsiveness of the isolated rat aorta and the mechanisms underlying this response.
Material and methods
Animals and experimental design
Male Wistar rats were housed under standard laboratory conditions with free access to food and water. The housing conditions and experimental protocols were approved by the Animal Ethics Committee of the University of São Paulo – Campus of Ribeirão Preto (# 11.1.1267.53.4).
Rats weighing 230–250 g (50 d old) were purchased from the animal facility of the University of São Paulo, Campus Ribeirão Preto. The rats were housed in groups of three per cage under a 12 h/12-h light/dark cycle (lights on at 06:30 h) at 23 ± 1 °C and given free access to Purina laboratory chow and water. The rats were randomly divided into two groups: control (n = 45) and stressed (n = 45). Rats from the stressed group were restrained for 2 h in a wire mesh chamber (6.3 × 19.3 cm) as previously described (Padovan & Guimarães, Citation2000). Rats were restrained always from 08:00 h to 10:00 h. Functional and biochemical analyses here described were conducted 24 h after the 2-h period of restraint. The restraint period was chosen based on previous findings showing that it induces anxiety-like effects 24 h later in rats (McBlane & Handley, Citation1994; Padovan & Guimarães, Citation2000). Control rats remained in their home cages.
Elevated plus-maze (EPM)
Rats from both groups were individually placed in the center of the EPM facing a closed arm and allowed 5 min of free exploration as previously described (Padovan & Guimarães, Citation2000). The EPM consisted of two open and two enclosed arms of equal length and width (50 × 10 cm). The open arms had a 1-cm high Plexiglas edge while the enclosed arms had 40-cm high wooden sides. Experiments were carried out in a sound-attenuated, temperature-controlled room, illuminated by two 40 W fluorescent lights placed 1.3 m away from the EPM (Padovan & Guimarães, Citation2000). The test in the EPM was carried out 24 h after the 2-h period of restraint. The number of entries with the four paws, and time spent in the open or enclosed arms of the EPM were recorded. The percent of open arm entries (100 × open/total entries) and of time spent in the open arms (100 open/open + enclosed) were calculated for each rat as standard anxiety indices. The total closed arm entries were calculated as a relative pure index of locomotor activity (Padovan & Guimarães, Citation2000).
Determination of plasma corticosterone concentration
Rats were anaesthetized with urethane 25% (1.25 g/kg, i.p., Sigma-Aldrich, St. Louis, MO) and decapitated. Trunk blood was collected in chilled tubes containing heparin (25 μl/ml of blood). Plasma was obtained after centrifugation (20 min, 1000 g, 4 °C) and stored at −80 °C. For measurements of plasma corticosterone a specific radioimmunoassay (RIA) was used after extraction with ethanol as described previously (Mecawi et al., Citation2013). The sensitivity of the RIA and the intra- and inter-assay coefficients of variation were 0.16 µg/dl and 5.1% and 8.1%. Results are expressed as µg/dl.
Blood pressure measurements
Systolic blood pressure was measured in conscious rats using a noninvasive tail-cuff plethysmograph (Plethysmograph EFF 306, Insight, Ribeirão Preto, Brazil). The rats were subjected to a period of adaptation to the plethysmograph across 4 d. During the adaptation period three consecutive measures (∼2 min apart) were obtained daily. The rats were maintained for 5–10 min in a warm chamber (37 ± 1 °C) and three consecutive recordings (∼2 min apart) were performed. Results are expressed as the mean of the three recordings. Tail-cuff recordings were obtained before or 2 h and 24 h after restraint. As tail-cuff measurement itself involves an acute restraint effect, rats used in this experimental protocol were not also subjected to behavioral tests in the EPM. Systolic blood pressure is expressed in mmHg.
Vascular reactivity experiments
Rats were anaesthetized with urethane 25% (1.25 g/kg, i.p., Sigma-Aldrich, St. Louis, MO) and then killed by decapitation. The thoracic aorta was removed, cleaned of adherent connective tissue, cut into rings (5–6 mm in length) and placed in 5-ml organ chambers containing Krebs solution as described previously (Yogi et al., Citation2012). The composition of the Krebs solution was as follows (in mM): NaCl, 118.0; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 15.0; glucose, 5.5 and CaCl2, 2.5. The rings were submitted to a basal tension of 1.5 g during a 60 min equilibration period, with the bath fluid being changed every 15–20 min. In some rings, the endothelium was removed mechanically by gently rolling on a thin wire inserted in the lumen of the vessel. Endothelial integrity was qualitatively assessed by measuring the degree of relaxation induced by acetylcholine (1 µM) in the presence of contractile tone induced by phenylephrine (0.1 µM). For studies of endothelium-intact vessels, the ring was discarded if the relaxation induced by acetylcholine was less than 50%. For studies of endothelium-denuded vessels, the rings were discarded if there was any degree of relaxation.
Cumulative concentration–response curves for phenylephrine (0.0001–10 μM) or serotonin (5-HT, 0.001–100 μM) were determined on endothelium-intact and endothelium-denuded aortic rings. Contraction is expressed as changes in the displacement (g) from baseline. In another set of experiments, aortic rings were pre-contracted with phenylephrine (0.1 μM) and when the contraction reached a plateau, acetylcholine (0.0001–10 μM) or sodium nitroprusside (SNP, 0.0001–0.3 μM) was added cumulatively. The magnitude of contraction induced by phenylephrine did not differ among the experimental groups at 0.1 μM; relaxation is expressed as a percentage change from the contraction induced at this phenylephrine concentration.
In order to investigate the mechanisms underlying the hyper-reactivity to phenylephrine, cumulative concentration–response curves for phenylephrine were obtained in endothelium-intact aortic rings from control and stressed rats after incubation for 30 min with the following drugs: NG-nitro-l-arginine methyl ester (l-NAME, non-selective NO synthase [NOS] inhibitor, 100 μM), 1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid (indomethacin, non-selective cyclooxygenase [COX] inhibitor, 10 μM), 6-isopropoxy-9-oxoxanthene-2-carboxylic acid (AH6809, antagonist of prostaglandin F2α [PGF2α] receptors, 10 μM), 7-[3-[[2-(phenylcarbamoyl)hydrazinyl]methyl]-7-oxabicyclo[2.2.1]heptan-2-yl]hept-5-enoic acid (SQ29548, antagonist of PGH2/thromboxane A2 [TXA2] receptors, 3 μM) or 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt monohydrate (tiron, superoxide anion scavenger [], 1 mM). All drugs were purchased from Sigma-Aldrich (St. Louis, MO). The agonist concentration–response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 3.0; GraphPad Software Inc., San Diego, CA). Agonist potencies and maximal responses were expressed as pD2 (negative logarithm of the molar concentration of agonist producing 50% of the maximal response) and Emax (maximum effect elicited by the agonist), respectively.
Determination of thromboxane B2 (TXB2) and prostaglandin F2α (PGF2α) in the rat aorta
The aortas were isolated and frozen in liquid nitrogen. The tissues were homogenized in enzyme immunoassay (EIA) buffer and centrifuged (2000 g, 15 min, 4 °C). The samples (50 µl) were deproteinized by precipitation using 50 µl of absolute ethanol kept at 4 °C, followed by stirring and remained for 30 min at −20 °C. The supernatant was centrifuged (4000 g, 10 min, 25 °C). TXB2, the stable metabolite of TXA2 and PGF2α were measured by EIA using commercially available kits #519031 (sensitivity: 50% B/B0: 60 pg/ml) and #516011 (sensitivity: 50% B/B0: 52 pg/ml), respectively (Cayman Chemical, Ann Arbor, MI). Results were normalized for protein concentration and are expressed as pg/mg protein. Protein concentrations in all experiments were determined using the method of Lowry (Bio-Rad Laboratories, Hercules, CA).
Determination of plasma thiobarbituric acid reactive substances (TBARS)
Plasma TBARS content was determined colorimetrically at 540 nm using a commercially available kit (#10009055, Cayman Chemical, Ann Arbor, MI). TBARS concentration was determined using a standard curve for malondialdehyde bis (MDA) (range of the kit: 0–50 nmol/ml). Results are expressed as nmol/ml of plasma.
Detection of aortic superoxide anions ( ) by lucigenin enhanced chemiluminescence
) by lucigenin enhanced chemiluminescence
The lucigenin-derived chemiluminescence assay was used to determine concentration in aortic homogenates as previously described (Yogi et al., Citation2012). Luminescence was measured in a luminometer (Orion II luminometer, Berthold Detection Systems, Pforzheim, Germany). Results are expressed as relative light unit (RLU)/mg protein.
Visualization of ROS generation with fluorescent dye dihydroethidium (DHE)
In situ production of ROS was visualized using DHE. Aortas were vertically embedded in Tissue-tek and sectioned transversely (5-μm-thick slices). The slices were incubated with DHE (10 µM, diluted in DMSO 0.01%) for 30 min, and then washed three times with cold phosphate buffered saline (PBS, pH 7.4) as previously described (Montenegro et al., Citation2011). Sections were examined by fluorescence microscopy (Leica Model SPE, Leica Imaging Systems Ltd., Wetzlar, Germany) using λex 405 nm laser excitation and the image was captured at ×400.
Detection of aortic H2O2 concentration
Amplex red (#A22188, Invitrogen, Carlsbad, CA) was used to measure aortic H2O2 concentration. The aorta was homogenized in Krebs solution [(mM): NaCl 130; KCl 4.7; KH2PO4 1.18; MgSO4 1.17; NaHCO3 14.9; Glucose 5.5; CaCl2 1.6; pH: 7.4] with a glass-to-glass homogenizer. The homogenates were centrifuged at 1000g (10 min, 4 °C). Fifty microliters of Amplex Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) and horseradish peroxidase type II (0.1 unit/ml) were added to the 50 μl of the samples. Samples were incubated (30 min) in the dark at room temperature (25 °C) in 96-well microplates. 10-Acetyl-3,7-dihydroxyphenoxazine is a colorless, non-fluorescent reagent that reacts with H2O2 to produce resorufin, a red-fluorescent compound that was analyzed using an excitation wavelength of 571 nm and an emission wavelength of 585 nm. Aortic H2O2 concentrations were calculated on the basis of an H2O2 standard curve. Results are expressed as µM/mg protein.
Measurement of plasma and aortic nitrate/nitrite (NOx)
The aortas were homogenized in 200 µl PBS buffer (pH 7.4) and centrifuged at 10,000 g (10 min, 4 °C). The supernatant was ultrafiltered at 14,000 g for 15 min at 24 °C using Amicon Ultra-0.5 ml 10 kDa (#UFC5010BK Amicon Ultra-0.5 ml 10 kDa, Millipore, Billerica, MA). NOx concentration was measured using a commercially available kit (#780001, Cayman Chemical, Ann Arbor, MI) (range of the kit: 0–50 µM). Results were normalized for protein concentration and are expressed as μmol/mg protein. To evaluate plasma NOx, the blood was collected with EDTA and centrifuged at 1000 g (20 min, 4 °C). Then, the samples were ultrafiltered (#UFC5010BK Amicon Ultra-0.5 ml 10 kDa, Millipore) to reduce absorbance due to the presence of hemoglobin in the samples. NOx was measured colorimetrically at 540 nm following recommendations for a commercially available kit (#780001, Cayman Chemical). Results are expressed as μmol/l of plasma.
Determination of total plasma antioxidant capacity
The total antioxidant capacity was measured as previously described (Gonzaga et al., Citation2014). Samples were analyzed according to recommendations for a commercially available kit (#709001, Cayman Chemical). Results are expressed as mM of antioxidant activity.
Determination of superoxide dismutase (SOD) and catalase (CAT) activity in the rat aorta
Aortas were homogenized in cold HEPES buffer (20 mM), pH 7.2, containing EGTA (1 mM), mannitol (210 mM) and sucrose (70 mM) per gram tissue. The SOD standard curve provides a means to quantify the activity of all three types of SOD (Cu/Zn-, Mn- and Fe-SOD). SOD activity was determined by a colorimetric method using a commercially available kit (kit #706002, Cayman Chemical, Ann Arbor, MI). One unit of SOD (U/ml) is defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radical.
CAT activity was determined in aortas by a colorimetric method using a commercially available kit (kit #707002, Cayman Chemical, Ann Arbor, MI). The method is based on the reaction of the CAT with methanol in the presence of an optimal concentration of H2O2. The formaldehyde produced is measured spectrophotometrically with 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole (Purpald®) as the chromogen. Purpald specifically forms a bicyclic heterocycle with aldehydes, which upon oxidation changes from colorless to a purple color, and absorbance is read at a wavelength of 540 nm. CAT activity is expressed in nmol/min/mg protein.
Determination of reduced glutathione (GSH) in the rat aorta
Aortic GSH was evaluated through its interaction with 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) as previously described (Gonzaga et al., Citation2014). Results are expressed as µgGSH/mg protein.
Western immunoblotting
Samples were prepared as previously described (Yogi et al., Citation2012). Forty micrograms of protein were separated by electrophoresis on a 10% polyacrylamide gel (1.5 h at 150 V) in mini-gel apparatus (Mini Protean III, Bio-Rad, Hercules, CA). Proteins were transferred onto a nitrocellulose membrane (1.5 h at 100 V). Nonspecific binding sites were blocked with skimmed milk (7%) in Tris-buffered saline solution (Tris-10 mM, NaCl-150 mM, Tween 20) for 1 h at room temperature. Membranes were incubated (overnight at 4 °C) with the following primary antibodies: COX-1 (1:1000, sc-166573, Santa Cruz Biotechnology, Dallas, TX), COX-2 (1:500, ab15191 Abcam, Cambridge, Cambridgeshire, UK), iNOS (1:500, sc-650, Santa Cruz Biotechnology) or eNOS (1:500, N3893, Sigma-Aldrich, St. Louis, MO). Signals were revealed with chemiluminescence after incubation with secondary antibodies for 90 min at room temperature. Signals were visualized using a ChemiDoc™ XRS+ (Bio-Rad) and quantified by densitometry. Beta-actin (1:5000, sc-4778, Santa Cruz Biotechnology) was used as an internal control.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Statistically significant differences were calculated by Student’s t-test or one-way ANOVA followed by Bonferroni’s multiple comparison test and p < 0.05 was considered as statistically significant.
Results
Effects of restraint stress on exploratory activity in the EPM and plasma corticosterone concentration
Rats from the stressed group showed a decreased percentage of entries (t(16) = 6.87, p < 0.05) and percentage of time spent in the open arms of the EPM (t(16) = 4.31, p < 0.05), as compared to control rats (). The number of entries into the enclosed arms did not differ between the groups (Control: 11.2 ± 1.4; n = 9; Stressed: 10.4 ± 1.7; n = 9). Restraint stress significantly increased plasma corticosterone concentration (t(13) = 3.30, p < 0.05), as compared to control rats ().
Figure 1. Effects of restraint stress on exploration of the elevated plus-maze (EPM), plasma corticosterone concentrations and systolic blood pressure. (A) The rats were tested in the EPM and results represent the percent of open arm exploration [% entries (open bars) and time spent (hatched bars) in open arms]. Values are mean ± SEM of n = 9 for each group. (B) Plasma corticosterone was evaluated by radioimmunoassay in control (n = 7) and stressed rats (n = 8). (C) Systolic arterial pressure was evaluated by plethysmography before or 2 and 24 h after restraint (n = 8 for each group). *Compared to control group (p < 0.05, Student’s t-test).
![Figure 1. Effects of restraint stress on exploration of the elevated plus-maze (EPM), plasma corticosterone concentrations and systolic blood pressure. (A) The rats were tested in the EPM and results represent the percent of open arm exploration [% entries (open bars) and time spent (hatched bars) in open arms]. Values are mean ± SEM of n = 9 for each group. (B) Plasma corticosterone was evaluated by radioimmunoassay in control (n = 7) and stressed rats (n = 8). (C) Systolic arterial pressure was evaluated by plethysmography before or 2 and 24 h after restraint (n = 8 for each group). *Compared to control group (p < 0.05, Student’s t-test).](/cms/asset/cb8e3535-c2c1-4c19-a707-058d367b939a/ists_a_1014790_f0001_b.jpg)
Effect of restraint stress on systolic blood pressure
Baseline values of systolic blood pressure were similar in rats from the control () and stressed groups. Immobilization of the rats for 2 h did not change systolic blood pressure. Similarly, no differences on blood pressure were found 24 h after the 2-h period of restraint ().
Effects of restraint stress on the aortic reactivity to phenylephrine, 5-HT, acetylcholine and SNP
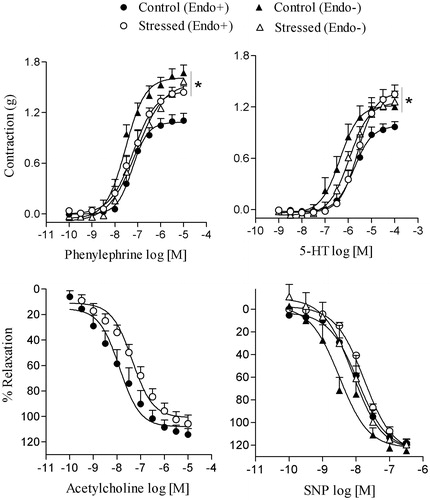
shows that restraint stress increased the maximal contraction (Emax) induced by phenylephrine (n = 8) in endothelium-intact rings, when compared to the control group (n = 7; F(4,27) = 10.38, p < 0.05); pD2 was not altered by stress (in both groups, pD2: 7.2 ± 0.1). In endothelium-denuded rings, the Emax or pD2 values for phenylephrine did not significantly differ between control (; pD2: 7.4 ± 0.1, n = 7) and stressed rats (pD2: 7.0 ± 0.2, n = 6). Similarly, Emax for 5-HT was increased after restraint stress in endothelium-intact (n = 5; ) but not endothelium-denuded aortic rings (n = 10), when compared to the control group with intact (n = 7), but not denuded endothelium (n = 6) (F(4,26) = 3.15, p < 0.05). Stress did not significantly alter pD2 for 5-HT (stress, intact, pD2: 5.6 ± 0.2; stress denuded, pD2: 6.0 ± 0.1; control, intact, pD2: 5.7 ± 0.1; control, denuded, pD2: 6.3 ± 0.2). Restraint stress did not alter Emax for the relaxation induced by acetylcholine (n = 13), when compared to the control group (n = 7; ); pD2 for acetylcholine was also not altered by stress (stress, pD2: 7.6 ± 0.2; control, pD2: 7.7 ± 0.3). Similarly, SNP-induced relaxation of endothelium-denuded rings was not different between control (n = 8) and stressed rats (n = 9), for Emax () (n = 9), and pD2 (control pD2: 8.4 ± 0.3; stress pD2: 8.2 ± 0.1). In endothelium-intact rings, the relaxation induced by SNP did not differ between control and stress groups for Emax or pD2 (control pD2: 8.0 ± 0.1, n = 6; stress pD2: 7.9 ± 0.1, n = 6) ().
Figure 2. Effects of restraint stress on the vascular reactivity of isolated aortas to phenylephrine, serotonin (5-HT), acetylcholine and sodium nitroprusside (SNP). The contraction induced by phenylephrine and 5-HT were determined in endothelium-intact (Endo+) and endothelium-denuded (Endo−) rat aortic rings. Relaxation to acetylcholine and SNP were obtained in Endo+ and Endo− aortic rings, respectively. Values are mean ± SEM of n = 6 for control and n = 9 for stressed rats. *Maximum effect elicited by the agonist (Emax) compared to control group Endo+ (p < 0.05, ANOVA followed by Bonferroni’s multiple comparison test).
Pre-incubation with l-NAME increased Emax for phenylephrine-induced contraction in endothelium-intact rings from control rats when compared to the contraction obtained in the absence of the inhibitor (F(3,28) = 3.34, p < 0.05) (; Supplementary Figure 1). Conversely, l-NAME did not alter Emax for phenylephrine in tissues from stressed rats when compared to the group without the inhibitor (). In the presence of indomethacin, the Emax values for phenylephrine in rings from stressed rats were reduced to values similar to the Emax in control aortas (F(3,26) = 5.68, p < 0.05). No changes in pD2 values were detected (). Similar results were found with tiron (F(3,29) = 7.11, p < 0.05) and SQ29548 (F(3,28) = 5.65, p < 0.05). However, AH6809 did not alter the contraction induced by phenylephrine in aortas from both control and stressed rats (F(3,29) = 6.01, p < 0.05) ().
Table 1. Effect of several inhibitors on the Emax (grams) and pD2 (−log EC50) values for contractile actions of phenylephrine on endothelium-intact rat aortic rings.
Effects of restraint stress on PGF2α and TXB2 concentration in the rat aorta
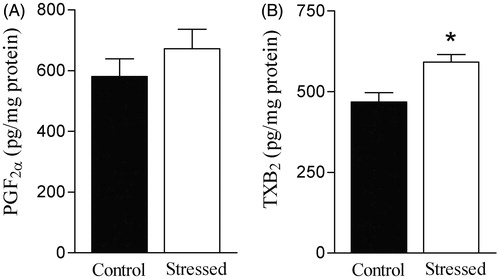
No difference in PGF2α concentration was found between aortas from control and stressed rats (). Restraint stress increased aortic TXB2 concentration (t(12) = 3.26, p < 0.05), when compared to the control group ().
Figure 3. Effects of restraint stress on prostaglandin (PG)F2α and thromboxane (TX)B2 concentrations in the rat aorta. Basal concentration of PGF2α (A) and TXB2 (B), a stable metabolite of TXA2, were measured in the rat aorta by enzyme immunoassay. Results are mean ± SEM of n = 6 for control and n = 7 for stressed rats. *Compared to control group (p < 0.05, Student’s t test).
Effects of restraint stress on generation, plasma TBARS and plasma and aortic nitrate/nitrite (NOx) concentration
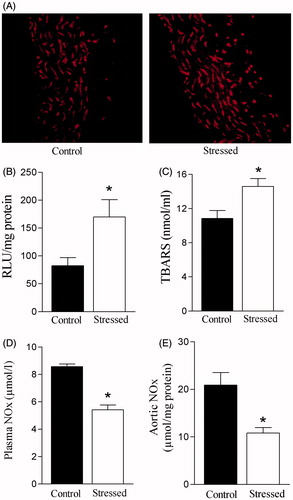
Restraint stress increased ROS generation in the rat aorta (). Lucigenin-derived luminescence was significantly greater (t(16) = 2.86, p < 0.05) in aortas from stressed rats (). Similarly, restraint stress increased plasma TBARS concentration (t(12) = 2.87, p < 0.05) (). Restraint stress significantly decreased basal plasma NOx concentration (t(14) = 7.88, p < 0.05) (). Similarly, aortic NOx concentration was decreased after restraint stress (t(14) = 3.54, p < 0.05) ().
Figure 4. Effects of restraint stress on systemic and vascular oxidative stress and basal nitrate/nitrite (NOx) levels. (A) Visualization of reactive oxygen species (ROS) generation in aorta slices detected with fluorescent dye dihydroethidium (DHE); note stronger signal after stress. (B) Bar graphs represent lucigenin-chemiluminescence in aortic tissue, (C) plasma concentrations of thiobarbituric acid reactive substances (TBARS). (D) Basal plasma, and (E) aortic nitrate/nitrite (NOx). Results are mean ± SEM of n = 7 for control and n = 11 for stressed rats. *Compared to control group (p < 0.05, Student’s t test).
Effect of restraint stress on the expression of NOS and COX isoforms in the rat aorta
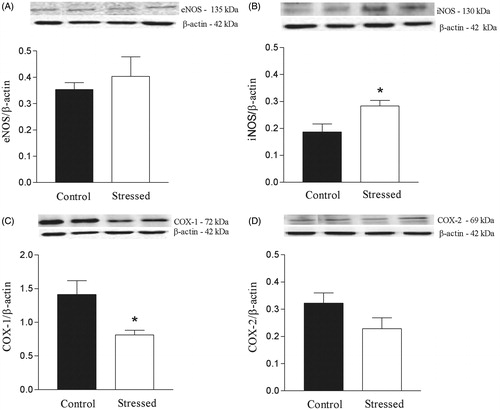
Restraint stress did not alter the expression of eNOS in the rat aorta (), but stress significantly increased the expression of iNOS (t(6) = 2.72, p < 0.05) (). Protein expression of COX-1, but not COX-2, was significantly decreased (t(6) = 2.73, p < 0.05) in aortas from stressed rats ().
Figure 5. Effects of restraint stress on endothelial NOS (eNOS), inducible NOS (iNOS), cyclooxygenase (COX)-1 and COX-2 expression in the rat aorta. Upper panels, representative immunoblots for eNOS, iNOS, COX-1 and COX-2 protein expression. Lower panels, corresponding bar graphs show densitometric data for expression of (A) eNOS, (B) iNOS, (C) COX-1 and (D) COX-2. Results are mean ± SEM of n = 4 for control and n = 6 for stressed rats. *Compared to control group (p < 0.05, Student’s t-test).
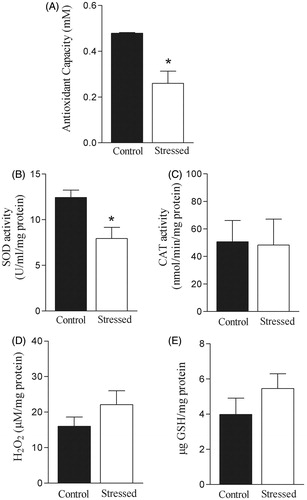
Effect of restraint stress on total plasma antioxidant capacity, SOD and CAT activities and H2O2 and GSH concentration in the rat aorta
Restraint stress reduced total plasma antioxidant capacity (t(14) = 4.07, p < 0.05) and SOD activity (t(10) = 3.05, p < 0.05) in the rat aorta (). No significant alterations in CAT activity or H2O2 and GSH concentrations in the aorta were observed after restraint stress ().
Figure 6. Effects of restraint stress on antioxidant system and hydrogen peroxide (H2O2) concentration. Total antioxidant capacity was evaluated in the plasma (A). Superoxide dismutase (SOD, B) and catalase (CAT) activities (C) as well as H2O2 (D) and reduced glutathione (GSH) concentration (E) were determined in the aorta. Results are presented as mean ± SEM of n = 6 for control and n = 8 for stressed rats. *Compared to control group (p < 0.05, Student’s t test).
Discussion
The present findings support our hypothesis that acute stress induces endothelial dysfunction, by a mechanism that involves increased generation of COX-derived vasoconstrictor prostanoids and oxidative stress. In the present study, restraint for 2 h induced a decrease in the exploration of the open arms of the EPM 24 h later, which is in accordance with previous findings describing that restraint induces anxiety-like effects in rodents (McBlane & Handley, Citation1994; Padovan & Guimarães, Citation2000). The restraint did not alter the number of enclosed arm entries, a parameter that reflects general exploratory activity; hence, our results indicate that the restraint stress did not result in a decreased general exploratory activity. Plasma corticosterone concentration, a biomarker of stress in rodents, is increased in rats submitted to restraint stress (Pellow et al., Citation1985; Pitman et al., Citation1988). Here, we found increased plasma corticosterone concentration 24 h after exposure to a 2 h-period of restraint. Taken together, our results indicate that restraint for 2 h induced anxiety-like and neuroendocrine effects which could be detected even 24 h later.
Restraint stress induces sustained blood pressure increases, which last through the restraint period (Crestani et al., Citation2010; Tavares & Correa, Citation2006). The lack of effect of restraint stress on blood pressure described here could be related to the period of restraint and the timing of blood pressure measurement. Hence, while we used a 2 h-period of restraint, Tavares & Correa (Citation2006) and Crestani et al. (Citation2010) restrained the rats for 60 and 30 min, respectively. Also, in those studies, a blood pressure increase was observed during restraint (Crestani et al., Citation2010; Tavares & Correa, Citation2006), and importantly, blood pressure returned to control values during post-stress recovery periods of 30 and 60 min (Crestani et al., Citation2010). This result indicates that the effect of acute restraint stress on blood pressure is transitory, which is in accordance with the present findings where no alteration in blood pressure was evidenced after the 2 h-period of restraint or 24 h later.
Alteration in the release of endothelial-derived mediators, such as NO and vasoconstrictor and/or vasodilator prostanoids, can lead to endothelial dysfunction, which is associated with increased vascular contractility (Tousoulis et al., Citation2014). In the present study, restraint stress increased the vascular contraction induced by phenylephrine and 5-HT in endothelium-intact, but not endothelium-denuded aortas, further indicating that acute restraint stress induces endothelial dysfunction. Indomethacin prevented the endothelium-dependent hyper-reactivity to phenylephrine in the aortas from stressed rats, further implicating endothelial vasoconstrictor prostanoids derived from COX in such a response. Increased concentration of TXB2, a stable metabolite of TXA2, was detected in aortas from stressed rats, which is consistent with the effect of the antagonist SQ29548 in the functional studies. Paradoxically, restraint stress decreased aortic COX-1 expression. COX-1 deficiency increases the expression of reciprocal and coupled enzymes such as phospholipase A2 and specific prostanoid synthases to overcome decreases in synthesis of prostanoids (Choi et al., Citation2006). Additionally, ROS increases COX-1 and COX-2 activity increasing the generation of contracting prostanoids (Virdis et al., Citation2007). Moreover, ROS increases TXA2 production and up-regulate thromboxane synthase expression (García-Redondo et al., Citation2009). Thus, increased COX-1 activity or increased expression of prostanoids synthases might compensate the reduction in COX-1 expression here described. Since no differences in COX-2 expression were observed, it is possible that this enzyme is not contributing to the increased generation of vasoconstrictor prostanoids in our model. Further studies are required to elucidate the role of COX isoforms in our model. Thus, our findings provide evidence that acute restraint stress increases the generation of endothelial prostanoids (possibly TXA2), which in turn increases vascular contractility.
Despite the endothelium-dependent increase in phenylephrine-induced contraction, acute stress did not alter the relaxation induced by acetylcholine, which is in accordance with previous findings in a model of chronic stress (Loria et al., Citation2011). A possible explanation for this effect is that differential regulation of endothelial NO release/generation may depend on the agonist producing endothelium-dependent relaxation, like acetylcholine, or contraction, like phenylephrine (Côrtes et al., Citation1996). Indeed, reduced endothelial-derived NO bioavailability is associated with endothelial dysfunction and increased vascular contractility (Tousoulis et al., Citation2014). Our results show that incubation of aortas from stressed rats with l-NAME did not alter phenylephrine-induced contraction. Importantly, endothelial NO counteracts phenylephrine-induced contraction in the rat aorta (Tirapelli et al., Citation2006). Hence, the increased responsiveness of aortas from stressed rats to phenylephrine here described could be a result of an impaired modulation of NO action on phenylephrine-induced contraction.
The isoform eNOS is constitutively expressed in the vasculature and increased contractility in response to phenylephrine is described when eNOS is down-regulated (Novella et al., Citation2013). This seems not to be the case in our study since no difference in eNOS expression was observed. The NO derived from iNOS reacts with to form peroxynitrite, an oxidant molecule (Xia & Zweier, Citation1997). For this reason, iNOS induction may be noxious and might ultimately lead to endothelial dysfunction. Thus, the increased aortic iNOS expression here described could be one mechanism by which acute stress induces endothelial dysfunction.
The present findings corroborate previous result describing that acute stress reduces plasma NO level (Yeh et al., Citation2002), and additionally show that acute stress reduced aortic concentration of NO. In general, reduced NO bioavailability may result from a decrease in NO synthesis or an increase in NO inactivation due to enhanced production. Our results show that acute stress increased
generation in the rat aorta. Importantly, the lucigenin-derived chemiluminescence assay used in the present study to access
production is based on the enzymatic action of the enzyme NAD(P)H oxidase (Pagano et al., Citation1995). Hence, the increased lucigenin-derived chemiluminescence here described indicates that the enzyme NAD(P)H oxidase is an important source of
generation by acute restraint stress in the vasculature. Of note, the finding that plasma TBARS concentration was increased in stressed rats indicates that increased ROS formation is probably a global phenomenon. Restraint stress also reduced the plasma antioxidant system, which is consistent with previous findings (Liu et al., Citation1994).
Of the ROS generated in vascular cells, and H2O2 appear to be especially important.
is considered as the primary ROS and it is described as inducing vascular contraction through increased concentration of intracellular calcium and generation of COX-derived vasoconstrictor prostanoids (Wong et al., Citation2010). In addition,
modulates the action of vasocontractile agents, such as phenylephrine in pathological conditions (Pernomian et al., Citation2012; Silva et al., Citation2013). Our functional studies demonstrated that
scavenging reduced the endothelium-dependent hyper-reactivity to phenylephrine induced by acute stress in endothelium-intact aortic rings. This observation supports the concept that
contributes to restraint stress-induced endothelium-dependent increase in vascular contraction. Superoxide anion may also react with NO to form peroxynitrite; this reaction reduces NO bioavailability leading to endothelial dysfunction and vasoconstriction (Queisser et al., Citation2010). This mechanism may also contribute to the endothelial dysfunction induced by acute restraint stress since reduced aortic NOx concentration was detected after stress. Thus, the endothelium-dependent hyper-reactivity to phenylephrine induced by acute stress may be related, at least in part, to an increased generation of
, which may serve to inactivate endothelial NO.
Superoxide anion is reduced by SOD to H2O2, which can be converted by CAT into H2O and O2. Both and H2O2 act as signaling molecules, but it is mainly H2O2 that is considered a signaling molecule because of its stability (Freinbichler et al., Citation2011). Depending on its concentration, H2O2 regulates signaling pathways that lead to vascular relaxation or contraction (Rodríguez-Martínez et al., Citation1998; Wei et al., Citation1996). Moreover, in the rat aorta H2O2 modulates the action of vasocontracting agents, such as phenylephrine (Silva et al., Citation2013). For those reasons, decreases or increases in H2O2 generation as well as alteration in the enzymes that regulate its concentration could be involved in the endothelium-dependent hyper-reactivity to phenylephrine here described after stress. Our findings show that acute stress decreased SOD activity without changing H2O2 concentration or CAT activity. This result indicates that H2O2 seems not to be involved in the endothelium-dependent hyper-reactivity to phenylephrine induced by acute stress. Mechanisms underlying the low SOD activity after stress are yet unclear. One possibility is post-translational protein modification by H2O2, which will mediate SOD inactivation (Hodgson & Fridovich, Citation1975; Jewett et al., Citation1999). Another possibility is SOD inactivation by
or peroxynitrite (Demicheli et al., Citation2007; MacMillan-Crow et al., Citation1998). Finally, despite the decrease in aortic SOD activity, no differences in GSH concentration were detected between the groups, suggesting that acute restraint stress differently modulates antioxidants systems in the tissue, as previously described in a model of acute immobilization stress (Liu et al., Citation1994).
Some limitations of this study should be pointed out. The use of a tail cuff for blood pressure measurements is a limitation as the procedure itself might induce acute stress. Moreover, since the increase in blood pressure induced by acute stress is transitory, detection of the effects of stress 2 and 24 h after restraint would be limited using plethysmography. Invasive blood pressure monitoring (e.g. radiotelemetry) would allow a more accurate evaluation of the short and long-term effects of acute stress. A second limitation is the use of aortic homogenates for biochemical analyses. These results will reflect the response of the whole aortic tissue to acute stress without distinction between endothelial and vascular smooth muscle cells.
The major new finding of our study is that acute stress induces endothelial-dependent exaggerated vascular contractility, by a mechanism that involves increased generation of COX-derived vasoconstrictor prostanoids and oxidative stress. Moreover, results presented here evidenced a reduction in the vascular antioxidant system. Although we have observed no effect of acute stress on arterial blood pressure, nonetheless acute stress induced vascular dysfunction, which has been regarded as the prognostic factor for predicting adverse cardiovascular events (Schachinger et al., Citation2000). In this regard, our data supports the notion that acute stress may contribute to development of cardiovascular diseases by inducing endothelial dysfunction.
Supplementary material available online
Supplementary Figure 1
Supplemental Material.pdf
Download PDF (113.1 KB)Acknowledgements
The authors thank Professor Francisco S. Guimarães for the use of a confocal microscope.
Declaration of interest
The authors report no conflicts of interest.
This work was supported by funds and grants from Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (#2013/04470-1 and #2013/00808-8). Dr. Mecawi is also supported by High Impact Research Chancellory Grant (UM.C/625/1/HIR/MOHE/MED/22 H-20001-E000086) from the University of Malaya.
References
- Bagchi D, Carryl OR, Tran MX, Bagchi M, Garg A, Milnes MM, Williams CB, et al. (1999). Acute and chronic stress-induced oxidative gastrointestinal mucosal injury in rats and protection by bismuth subsalicylate. Mol Cell Biochem 196(1–2):109–16
- Bosma H, Marmot MG, Hemingway H, Nicholson AC, Brunner E, Stansfeld SA. (1997). Low job control and risk of coronary heart disease in Whitehall II (prospective cohort) study. BMJ 314:558–65
- Choi SH, Langenbach R, Bosetti F. (2006). Cyclooxygenase-1 and -2 enzymes differentially regulate the brain upstream NF-kappa B pathway and downstream enzymes involved in prostaglandin biosynthesis. J Neurochem 98(3):801–11
- Chung IM, Kim YM, Yoo MH, Shin MK, Kim CK, Suh SH. (2010). Immobilization stress induces endothelial dysfunction by oxidative stress via the activation of the angiotensin II/its type I receptor pathway. Atherosclerosis 213(1):109–14
- Côrtes SF, Andriantsitohaina R, Stoclet JC. (1996). Alterations of cyclo-oxygenase products and NO in responses to angiotensin II of resistance arteries from the spontaneously hypertensive rat. Br J Pharmacol 119(8):1635–41
- Crestani CC, Tavares RF, Alves FH, Resstel LB, Correa FM. (2010). Effect of acute restraint stress on the tachycardiac and bradycardiac responses of the baroreflex in rats. Stress 13(1):61–72
- Demicheli V, Quijano C, Alvarez B, Radi R. (2007). Inactivation and nitration of human superoxide dismutase (SOD) by fluxes of nitric oxide and superoxide. Free Radic Biol Med 42(9):1359–68
- Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Dawood T. (2008). Chronic mental stress is a cause of essential hypertension: presence of biological markers of stress. Clin Exp Pharmacol Physiol 35(4):498–502
- Freinbichler W, Colivicchi MA, Stefanini C, Bianchi L, Ballini C, Misini B, Weinberger P, et al. (2011). Highly reactive oxygen species: detection, formation, and possible functions. Cell Mol Life Sci 68:2067–79
- García-Redondo AB, Briones AM, Beltrán AE, Alonso MJ, Simonsen U, Salaices M. (2009). Hypertension increases contractile responses to hydrogen peroxide in resistance arteries through increased thromboxane A2, Ca2+, and superoxide anion levels. J Pharmacol Exp Ther 328:19–27
- Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O'Connor G, et al. (2000). Mental stress induces transient endothelial dysfunction in humans. Circulation 102(20):2473–8
- Gonzaga NA, Callera GE, Yogi A, Mecawi AS, Antunes-Rodrigues J, Queiroz RH, Touyz RM, Tirapelli CR. (2014). Acute ethanol intake induces mitogen-activated protein kinase activation, platelet-derived growth factor receptor phosphorylation, and oxidative stress in resistance arteries. J Physiol Biochem 70(2):509–23
- Grimée R, Wülfert E. (1995). Acute stress in rats produces a rapid and sustained increase in prostacyclin production in aortic tissue: dependence on corticosterone. Life Sci 57(1):69–81
- Hodgson EK, Fridovich I. (1975). The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme. Biochemistry 14:5294–9
- Jewett SL, Rocklin AM, Ghanevati M, Abel JM, Marach JA. (1999). A new look at a time-worn system: oxidation of CuZn-SOD by H2O2. Free Radic Biol Med 26(7–8):905–18
- Liu J, Mori A. (1994). Involvement of reactive oxygen species in emotional stress: a hypothesis based on the immobilization stress-induced oxidative damage and antioxidant defense changes in rat brain, and the effect of antioxidant treatment with reduced glutathione. Int J Stress Manag 1(3):249–63
- Liu J, Wang X, Mori A. (1994). Immobilization stress-induced antioxidant defense changes in rat plasma: effect of treatment with reduced glutathione. Int J Biochem 26(4):511–17
- Loria AS, Kang KT, Pollock DM, Pollock JS. (2011). Early life stress enhances angiotensin II-mediated vasoconstriction by reduced endothelial nitric oxide buffering capacity. Hypertension 58(4):619–26
- MacMillan-Crow LA, Crow JP, Thompson JA. (1998). Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 37:1613–22
- Manni L, Antonelli A, Costa N, Aloe L. (2005). Stress alters vascular-endothelial growth factor expression in rat arteries: role of nerve growth factor. Basic Res Cardiol 100(2):121–30
- Matsumoto K, Yobimoto K, Huong NT, Abdel-Fattah M, Van Hien T, Watanabe H. (1999). Psychological stress-induced enhancement of brain lipid peroxidation via nitric oxide systems and its modulation by anxiolytic and anxiogenic drugs in mice. Brain Res 839(1):74–84
- McBlane JW, Handley SL. (1994). Effects of two stressors on behaviour in the elevated X-maze: preliminary investigation of their interaction with 8-OH-DPAT. Psychopharmacology 116:173–82
- McDougall SJ, Lawrence AJ, Widdop RE. (2005). Differential cardiovascular responses to stressors in hypertensive and normotensive rats. Exp Physiol 90(1):141–50
- Mecawi AS, Vilhena-Franco T, Fonseca FV, Reis LC, Elias LL, Antunes-Rodrigues J. (2013). The role of angiotensin II on sodium appetite after a low-sodium diet. J Neuroendocrinol 25(3):281–91
- Montenegro MF, Amaral JH, Pinheiro LC, Sakamoto EK, Ferreira GC, Reis RI, Marçal DM, et al. (2011). Sodium nitrite downregulates vascular NADPH oxidase and exerts antihypertensive effects in hypertension. Free Radic Biol Med 51:144–52
- Neves VJ, Moura MJ, Tamascia ML, Ferreira R, Silva NS, Costa R, Montemor PL, et al. (2009). Proatherosclerotic effects of chronic stress in male rats: altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress 12(4):320–7
- Novella S, Dantas AP, Segarra G, Vidal-Gómez X, Mompeón A, Garabito M, Hermenegildo C, Medina P. (2013). Aging-related endothelial dysfunction in the aorta from female senescence-accelerated mice is associated with decreased nitric oxide synthase expression. Exp Gerontol 48(11):1329–37
- Okruhlicová L, Dlugosová K, Mitasíková M, Bernátová I. (2008). Ultrastructural characteristics of aortic endothelial cells in borderline hypertensive rats exposed to chronic social stress. Physiol Res 57(Suppl 2):S31–7
- Padovan CM, Guimarães FS. (2000). Restraint-induced hypoactivity in an elevated plus-maze. Braz J Med Biol Res 33(1):79–83
- Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA. (1995). An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol 268(6 Pt 2):H2274–80
- Pellow S, Chopin P, File SE, Briley M. (1985). Validation of open: closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods 14(3):149–67
- Pernomian L, Gomes MS, Restini CA, Ramalho LNZ, Tirapelli CR, de Oliveira AM. (2012). The role of reactive oxygen species in the modulation of the contraction induced by angiotensin II in carotid artery from diabetic rat. Eur J Pharmacol 678(1–3):15–25
- Persoons JH, Schornagel K, Brevé J, Berkenbosch F, Kraal G. (1995). Acute stress affects cytokines and nitric oxide production by alveolar macrophages differently. Am J Respir Crit Care Med 152(2):619–24
- Pitman DL, Ottenweller JE, Natelson BH. (1988). Plasma corticosterone levels during repeated presentation of two intensities of restraint stress: chronic stress and habituation. Physiol Behav 43(1):47–55
- Queisser N, Fazeli G, Schupp N. (2010). Superoxide anion and hydrogen peroxide-induced signaling and damage in angiotensin II and aldosterone action. Biol Chem 391:1265–79
- Rodríguez-Martínez MA, García-Cohen EC, Baena AB, González R, Salaíces M, Marín J. (1998). Contractile responses elicited by hydrogen peroxide in aorta from normotensive and hypertensive rats: endothelial modulation and mechanism involved. Br J Pharmacol 125:1329–35
- Schachinger V, Britten MB, Zeiher AM. (2000). Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 101(16):1899–906
- Silva BR, Pernomian L, Grando MD, Amaral JH, Tanus-Santos JE, Bendhack LM. (2013). Hydrogen peroxide modulates phenylephrine-induced contractile response in renal hypertensive rat aorta. Eur J Pharmacol 721(1–3):193–200
- Tavares RF, Correa FM. (2006). Role of the medial prefrontal cortex in cardiovascular responses to acute restraint in rats. Neuroscience 143(1):231–40
- Tirapelli CR, Al-Khoury J, Bkaily G, D’Orléans-Juste P, Lanchote VL, Uyemura SA, de Oliveira AM. (2006). Chronic ethanol consumption enhances phenylephrine-induced contraction in the isolated rat aorta. J Pharmacol Exp Ther 316(1):233–41
- Tousoulis D, Simopoulou C, Papageorgiou N, Oikonomou E, Hatzis G, Siasos G, Tsiamis E, Stefanadis C. (2014). Endothelial dysfunction in conduit arteries and in microcirculation. Novel therapeutic approaches. Pharmacol Ther 144(3):253–67
- Virdis A, Colucci R, Fornai M, Duranti E, Giannarelli C, Bernardini N, Segnani C, et al. (2007). Cyclooxygenase-1 is involved in endothelial dysfunction of mesenteric small arteries from angiotensin II-infused mice. Hypertension 49(3):679–86
- Wei EP, Kontos HA, Beckman JS. (1996). Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol 271(3 Pt 2):H1262–6
- Wong MS, Delansorne R, Man RY, Svenningsen P, Vanhoutte PM. (2010). Chronic treatment with vitamin D lowers arterial blood pressure and reduces endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 299(4):H1226–34
- Xia Y, Zweier JL. (1997). Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA 94:6954–8
- Yeh CB, Leckman J, Wan FJ, Shiah IS, Lu RB. (2002). Characteristics of acute stress symptoms and nitric oxide concentration in young rescue workers in Taiwan. Psychiatry Res 112(1):59–68
- Yogi A, Callera GE, Mecawi AS, Batalhão ME, Carnio EC, Antunes-Rodrigues J, Queiroz RH, et al. (2012). Acute ethanol intake induces superoxide anion generation and mitogen-activated protein kinase phosphorylation in rat aorta: a role for angiotensin type 1 receptor. Toxicol Appl Pharmacol 264(3):470–8