
Abstract
The last decade has witnessed profound growth in studies examining the role of fundamental neuroimmune processes as key mechanisms that might form a natural bridge between normal physiology and pathological outcomes. Rooted in core concepts from psychoneuroimmunology, this review utilizes a succinct, exemplar-driven approach of several model systems that contribute significantly to our knowledge of the mechanisms by which neuroimmune processes interact with stress physiology. Specifically, we review recent evidence showing that (i) stress challenges produce time-dependent and stressor-specific patterns of cytokine/chemokine expression in the CNS; (ii) inflammation-related genes exhibit unique expression profiles in males and females depending upon individual, cooperative or antagonistic interactions between steroid hormone receptors (estrogen and glucocorticoid receptors); (iii) adverse social experiences incurred through repeated social defeat engage a dynamic process of immune cell migration from the bone marrow to brain and prime neuroimmune function and (iv) early developmental exposure to an inflammatory stimulus (carageenin injection into the hindpaw) has a lasting influence on stress reactivity across the lifespan. As such, the present review provides a theoretical framework for understanding the role that neuroimmune mechanisms might play in stress plasticity and pathological outcomes, while at the same time pointing toward features of the individual (sex, developmental experience, stress history) that might ultimately be used for the development of personalized strategies for therapeutic intervention in stress-related pathologies.
Introduction
Few experiences are more common among people than exposure to stressful life events, with stress provoking situations appearing to permeate the lives of many on a daily basis. On one end of the spectrum, people often endure a seemingly perpetual range of minor, daily events (or hassles) that aggregate into more profound chronic periods of distress. Other stressful experiences arise in the form of severe, but temporally brief, threats that can instill life-long changes in psychological outlook and stress physiology. Consistent with this, 22% of Americans reported extreme stress in their daily lives, with as much as 44% of Americans reporting that their stress had increased in the past 5 years (APA, 2012). Beyond the momentary discomfort incurred by stressful life experiences, decades of biomedical research have established positive associations between life stress and the development of a wide range of pathological disease states, including psychiatric disorders such as Major Depressive Disorder (Maes et al., Citation2012; Weidenfeld & Yirmiya, Citation1996) and Post-traumatic Stress Disorder (Pace & Heim, Citation2011; Yehuda et al., Citation2009); neurodegenerative disorders such as Alzheimers disease (Jeong et al., Citation2006) and Parkinson’s disease (Macht et al., Citation2005); systemic health disorders such as cardiovascular disease (Black & Garbutt, Citation2002), diabetes, and other metabolic disorders (Cohen et al., Citation2010), and inflammatory diseases (Elenkov & Chrousos, Citation2002; Sternberg, Citation2001). These findings underscore the importance of developing highly tractable, preclinical stress models that can delineate complex interactions among various, stress-responsive physiological systems.
Multiple physiological systems are activated in response to stressful experiences and these systems are highly conserved across species. Most typically, the sympathetic nervous system (SNS) and hypothalamic–pituitary–adrenal (HPA) axis are thought to be key drivers of many downstream physiological effects of stress. These systems undoubtedly play a key role in orchestrating comprehensive, organism-wide strategies to promote survival in the face of threats that might be physiological (hypoglycemia, hemorrhage, injury, infection, etc.) or psychological (predator exposure, social dominance, startle, etc.) in nature. The immune system, however, is a prime example of another physiological system that is profoundly impacted by stressful experiences, with acute and chronic stress often producing differential immunological outcomes (Dhabhar et al., Citation1995). Indeed, a wealth of studies have examined the impact of stress on immune function and infection vulnerability (Deak et al., Citation1999), and one prevailing principle has been the role of bidirectional communication between the brain and the immune system (Maier & Watkins, Citation1998). Bidirectional communication involves efferent and afferent pathways by which the brain and periphery exchange information about the homeostatic state of the organism, and is an essential element of a mammal’s response to environmental, physiological and psychological stressors that disrupt homeostasis. Depending on the stressor (i.e. the nature, the intensity and the duration of the threat), neuroendocrine modulation of immunity may be beneficial or detrimental (Anisman, Citation2009; Dantzer et al., Citation2008). For instance, immune dysfunction and behavioral disturbances have been associated with chronic or repeated exposure to stress, and elevated levels of circulating proinflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) have been associated with mood disturbances such as anxiety and depression (Miller, Citation2008; Pace et al., Citation2006). Furthermore, stressful experiences can induce activation of many aspects of both peripheral immunity and central neuroimmune processes, both of which contribute to various forms of host defense, recovery from stress and ultimately disease susceptibility. It is this latter aspect of stress–immune interactions, namely neuroimmune consequences of stress, which has been the subject of intense work by multiple laboratories in recent years and is the primary emphasis of the present review.
Neuroimmune consequences of stress and implications for elaboration of the stress response
To better understand neuroimmune consequences and mechanisms of stress, it seems prudent to acknowledge first the multitude of cellular changes that might constitute neuroimmune or neuroinflammatory processes. Perhaps the most commonly studied are those that involve the expression of cytokines, chemokines and/or other molecules known to invoke (or in some cases, dampen) immunological processes. For example, numerous studies from our lab and others have demonstrated that interleukin-1β (IL-1) was rapidly increased in key limbic structures (paraventricular nucleus of the hypothalamus, PVN; amygdala) in response to stress challenges that involve application of an aversive/noxious stimulus such as footshock (Blandino et al., Citation2009; Deak et al., Citation2003; Hueston & Deak, Citation2014; Nguyen et al., Citation1998), but not in response to social stress challenges (Hueston et al., Citation2011). In contrast, social stressors appear to increase release of another pro-inflammatory cytokine, interleukin-6 (IL-6), in both plasma and brain of females (Hodes et al., Citation2014). Although IL-1, IL-6, and TNF-α are considered to be the classic proinflammatory cytokines – and all appear to be modulated in the CNS by stressor exposure – these are by no means the only neuroimmune factors impacted by stress. For example, monocyte chemoattractive protein-1 (MCP1/CCL2) is a chemokine expressed in the CNS in response to a number of stress challenges and likely plays a key role in recruitment of monocytes from the blood into the CNS (Blandino et al., Citation2009; Wohleb et al., Citation2013; as reviewed further below). Prostaglandins also appear to be highly active in cortical regions after stress (Garcia-Bueno et al., Citation2008). Thus, a multitude of distinct neuroimmune signaling pathways appear to be activated in response to stressful circumstances, and these effects often depend upon specific individual subject characteristics such as sex, age, strain and prior stress history (for a recent review, see Hueston & Deak, Citation2014).
In addition to stress-related increases in expression of cytokines and other inflammatory signaling molecules, stress exposure is often accompanied by other cellular manifestations of neuroimmune activation, such as dynamic changes in microglial activation state. Microglia play a prominent role in immune surveillance within the CNS, and are often viewed as the “first responders” in response to infection and/or damage. Prior studies have shown that microglia are highly responsive to stress, with evidence to date suggesting that microglia exhibit priming-like effects following stress exposure as evidenced by enhanced or accelerated activational responses to subsequent immune challenge either in vivo (Johnson et al., Citation2002a,Citationb) or ex vivo (Frank et al., Citation2010). An enhanced functional activation state of microglia is also associated with altered expression of cell surface markers that are known to modify or regulate microglia reactivity to antigens (Blandino et al., Citation2009; Frank et al., Citation2007). Other studies have shown that chronic stress enhanced microglial proliferation (Nair & Bonneau, Citation2006) and migration into the CNS (Wohleb et al., Citation2013; discussed in greater detail below). Although it is not clear whether these changes in microglia are the cause or consequence of cytokine expression, it is clear that microglial activation is associated with increased cytokine expression, and exogenous cytokine administration often enhances microglial activation. In this sense, it is notable that IL-1 in particular is often described in the neuroinjury literature as an immediate early gene indicative of microglial activation state. Regardless, the auto-induction of neuroimmune signaling pathways underscores the positive feedback trajectory often exhibited by the immune system to rapidly squelch replicating pathogens and mitigate damage.
Together, these findings provoke several very interesting questions that remain unresolved, including (i) Are there unique, stressor-specific cytokine response profiles that might indicate a stress-selective cytokine signature?; (ii) Do cytokine responses in the CNS occur in a brain site-selective manner, or do these changes occur across more gross functional regions of the CNS as is typical for innate, immunologically-based challenges such as bacterial infection?; (iii) What is the relationship between expression of a single cytokine (versus the cytokine milieu), the mechanisms which drive those cytokine changes, and the ultimate impact on CNS function (cognitive, behavioral, or neuropathological)? and (iv) What are the natural mechanisms which limit, shut off, or otherwise constrain neuroimmune consequences of stress? The answers to these and many other questions remain to be determined, and recent advances in the availability of multiplex assays with high sensitivity for detection of inflammatory molecules will be valuable tools for answering them in the near future.
In the mean time, substantial progress has been made in demonstrating the functional impact of stress-dependent expression of cytokines. For instance, administration of specific pathway inhibitors such as IL-1 receptor antagonist (intra-cerebroventricular; i.c.v.) has been shown to block behavioral deficits indicative of learned-helplessness (Maier & Watkins, Citation1995), and the enhanced HPA axis response produced by inescapable tailshock (Johnson et al., Citation2002a). In our hands, IL-1 receptor antagonist reversed the suppression of social interaction produced by prior stressor exposure, and blocked the release of aversive odor cues emitted by stressed rats (Arakawa et al., Citation2009). Agents with more global anti-inflammatory properties, such as alpha-melanocyte stimulating hormone (α-MSH), have been shown to rescue stress-induced reductions in food intake and HPA axis aberrations (Milligan et al., Citation1998). IL-10, a potent anti-inflammatory cytokine, has also been shown to reverse sickness-like behavioral responses in a guinea pig model of maternal separation (Hennessy et al., Citation2011), and sensitization of febrile responses produced by this maternal separation experience was reversed with naproxen, a relatively selective inhibitor of cyclooxygenase-2 activity (i.e. the enzyme responsible for production of mature prostaglandin E2 synthesis) – a final common mediator of many inflammation-related effects (Hennessy et al., Citation2015). Although there is some recent evidence suggesting that antagonists to the P2X7 (purinergic) receptor can reverse some stress-related effects (presumably due to blocking IL-1 release), these effects are generally modest, perhaps because they are blocking only one cytokine within the context of many (Catanzaro et al., Citation2014; Lord et al., Citation2014). Thus, one very fruitful avenue of future studies will be the development of novel therapeutics targeting inflammatory signaling pathways that can be used to ameliorate adverse health consequences of stress. The key challenge for production of these therapeutics will be the formulation of agents that either (a) readily cross the blood-brain barrier (BBB) or (b) can be coupled to some form of vector (viruses, nanoparticles, etc.) in order to promote CNS penetration of the drugs.
Mechanisms controlling stress-dependent changes in neuroimmune function
Having described a number of typical neuroimmune alterations evoked by stress challenges, we now shift our attention to the mechanisms that appear to be driving these neuroimmune changes. Here, we entertain the possibility that multiple, distinct pathways lie upstream of stress-dependent activation of neuroimmune function, and that these different pathways might be clustered into specific genres. Chief among these mechanisms is the role that rapid neural signals might play in the initiation of stress-dependent neuroimmune processes. Perhaps the best studied of these neural signals is the role of norepinephrine (NE) due to its primary role as a stress hormone. A multitude of studies have shown inhibiting NE signaling via functional antagonist administration seems to block many cytokine changes evoked by stress. Interestingly, central expression of IL-1 was blocked by pretreatment with propranolol (a beta-adrenergic antagonist), whereas plasma cytokine responses were unaffected by this drug. In contrast, administration of an alpha1 adrenergic antagonist (prazosin) blocked the plasma IL-1 response, but had no effect on central cytokine expression in the same subjects (Johnson et al., Citation2005). Consistent with these findings, pretreatment with desipramine increased both basal and stress-dependent expression of IL-1 (Blandino et al., Citation2006, Citation2009), and isoproterenol recapitulated the effects of stress by inducing central expression of IL-1. Further studies showed that i.c.v. administration of the catecholamine-selective neurotoxin, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP4), blocked stress-dependent IL-1 expression in the hippocampus (Johnson et al., Citation2005), whereas more focal lesions of the ventral noradrenergic bundle (via 6-hydroxydopamine; 6-OHDA) produced a much more modest effect on IL-1 expression in the PVN (Blandino et al., Citation2013). Overall, these data provide a compelling role for NE signaling in the expression of at least some neuroimmune consequences of stress. However, in other studies, chronic administration of MK-801 blocked microglial proliferative responses in brain (Nair & Bonneau, Citation2006), suggesting that multiple excitatory transmitters may contribute to neuroimmune responses to stress. Considering that ATP is co-packaged and co-released from sympathetic nerve terminals with NE, it is perhaps not surprising that ATP and its derivatives are now being considered as moderators of neuroimmune processes as well (Catanzaro et al., Citation2014; Lord et al., Citation2014).
In addition to these rapid neural signals that may promote neuroimmune responses to stress, emerging evidence suggests alternative pathways involving danger, damage, and disease-associated signals as key drivers. The basic premise here is that signals originating within the host organism, termed Danger Associated Molecular Patterns (DAMPs), are released in response to threatening stimuli (or overt tissue damage) and interact with Pathogen Recognition Receptors, which are positively coupled to inflammatory signaling pathways. Some examples of DAMPs include high mobility group box 1 (Weber et al., Citation2015) and heat shock proteins (Hsp), particularly Hsp72 (Johnson & Fleshner, Citation2006). These molecules are typically released in response to cellular damage and serve as co-stimulatory molecules that can interact with the same receptors known to recognize nonspecific molecular motifs expressed by classes of bacteria and viruses. In addition, ongoing studies are examining the impact of stress exposure on the gut microbiome, which may either directly or indirectly influence cytokine expression or other aspects of neuroimmune function through translocation of bacteria across the gastrointestinal tract (Bailey et al., Citation2011; Mayer et al., Citation2014). Finally, there is scattered evidence to suggest that cytokines (and other inflammatory signaling molecules) may be expressed in very low quantities under ambient conditions, and might be released as initiators of downstream signaling processes, including auto-induction. This hypothesis is supported by the observation of certain rapid changes involving cytokines that are probably too quick to involve de novo synthesis of peptide, and the fact that a variety of inflammatory-related disease states are associated with enhanced HPA axis output (in the absence of canonical endocrine signals; e.g. Goshen et al., Citation2003).
Critical to the discussion of signals that drive neuroimmune consequences of stress is the discussion of how such neuroimmune processes are constrained later in the stress response cycle (Deak, Citation2007). Classically speaking, glucocorticoids (GCs) have been regarded as potently anti-inflammatory, and there is strong evidence to support the view that GCs effectively constrain neuroimmune consequences of stress as well. Initial studies examining IL-1 protein expression in brain after tailshock required that rats be adrenalectomized (ADX) in order to unmask the IL-1 response (Nguyen et al., Citation1998) – an effect that was normalized by exogenous corticosterone administration to ADX rats exposed to stress (Nguyen et al., Citation2000). As extraction procedures and assay sensitivity have improved dramatically over the years, subsequent studies demonstrated increased expression of IL-1 even in adrenal intact rats (e.g. Deak et al., Citation2003, Citation2005), with ADX or administration of the GC synthesis inhibitor, metyrapone, still potentiating the IL-1 response to stress (Blandino et al., Citation2006, Citation2009). Interestingly, it seems that the role of corticosterone as an endogenous inhibitor of inflammation may switch toward pro-inflammatory based on the dose and/or timing of corticosterone release (Frank et al., Citation2013, Citation2014; Sorrells & Sapolsky, Citation2007). These new findings do not negate the overwhelming amount of data supporting the role of GCs as principally anti-inflammatory in nature, but instead add a novel twist of intrigue to the many layers of neural and hormonal interactions that seem to regulate – and counter-regulate – neuroimmune responses to stress. Consistent with this, it can be noted that the clinical use of corticosteroids to ameliorate inflammatory-related conditions typically involves doses that achieve plasma concentrations of GCs that are orders of magnitude larger than is possible for endogenous concentrations of GCs.
When considering the role of GCs on neuroimmune consequences of stress within a normal physiological context, one must consider other complex aspects of physiology that may influence stress reactivity and/or neuroimmune consequences of stress. For instance, activation of the HPA axis does not lead to the sole release of GC hormone from the adrenal gland, but instead leads to the release of a multitude of other hormonal mediators, many of which have immunomodulatory properties (Deak, Citation2008). Notably, progesterone is a precursor steroid that is enzymatically and rapidly converted to corticosterone in the adrenal glands, and yet plasma progesterone evinces a rapid surge that parallels plasma corticosterone in both male (Hueston & Deak, Citation2014) and female (Arakawa et al., Citation2014) rats. Additionally, there are virtually no manipulations that adequately discriminate between corticosterone and progesterone action since metyrapone administration or ADX ablate both corticosterone and progesterone (Hueston & Deak, Citation2014), and most GC-selective antagonists do not adequately discriminate between corticosteroid and progestin receptors (Etgen & Barfield, Citation1986; Rupprecht et al., Citation1993). This is relevant in the present context because progesterone has also been shown to have anti-inflammatory effects (particularly at high doses; Stein, Citation2008; van der Burg & van der Saag, Citation1996), and thus the attribution of an anti-inflammatory role of corticosterone during times of stress may be circumspect. Future studies should seek to be more discerning about the relative influence of corticosterone and progesterone as moderators of neuroimmune processes.
Although very few studies have addressed sex differences in neuroimmune responses to stress, our lab recently completed a series of studies showing that ovariectomy led to (i) increased basal and stress-evoked expression of IL-1 in the PVN relative to sham-operated females, and that (ii) this effect was recovered by replacement of estradiol (Arakawa et al., Citation2014). Although we also observed some fluctuation in stress-dependent expression of IL-1 across the ovarian cycle (i.e. the IL-1 response in the PVN was less pronounced during metestrus), this latter effect requires additional follow-up studies to better understand the mechanism. Furthermore, it should be noted also that the primary neuroimmune measure in this series of studies was IL-1 gene expression, and ongoing studies are employing a more comprehensive analysis of neuroimmune changes to better understand how neuroimmune consequences and mechanisms of stress might be sex-dependent. The role that androgens and estrogens play in the regulation of inflammation is indeed quite complex, and the direction of the interactions often vary as a function of developmental epoch (puberty versus adulthood versus aging) and the complex physiological milieu incurred by the stress challenge at hand. In the section that follows, we take much deeper consideration of sex differences in inflammatory processes, and how steroid hormone signaling exhibits unique, overlapping, cooperative and antagonistic effects on inflammatory gene regulation.
GC receptor/estrogen receptor crosstalk: implications for the sexually dimorphic actions of GCs
Both the adaptive and maladaptive responses to stress are mediated via GCs binding to the intracellular glucocorticoid receptor (GR), a ligand activated transcription factor. GR is ubiquitously expressed and regulates an array of physiological processes, most notably regulation of inflammation and metabolism (Oakley & Cidlowski, Citation2013). While stress has been well established as a risk factor for a variety of pathologies ranging from metabolic abnormalities to cardiovascular disease and even various psychiatric disorders, it is now appreciated that GCs serve more than just mediators of the stress response as global deletion of GR leads to post-natal lethality, indicating the importance of GCs for survival (Schmid et al., Citation1995). As a ligand-activated transcription factor, GR is a member of the nuclear receptor superfamily of proteins. In the absence of ligand, GR is sequestered in the cytoplasm in a multi-protein complex including chaperone proteins such as heat shock protein 90 (Hsp90). Once bound to ligand, GR undergoes a conformational change resulting in the dissociation from the chaperone protein complex and allowing for its nuclear translocation. In the nucleus GR can regulate gene expression through a variety of mechanisms. The most classical and well-studied mode of gene regulation is the direct binding of GR to specific DNA sequences, termed glucocorticoid response elements, leading to either transactivation or transrepression. An alternative mode of action is not by binding directly to DNA, but rather tethering to other transcription factors such as AP-1 or NFκ-B (Oakley & Cidlowski, Citation2011).
Sexual dimorphism
As the stress response is part of normal biology, sexual dimorphism is also a key component of biology. Sexual dimorphism refers to sex-related phenotypic differences that are due to the varying genetic landscape between males and females (Quinn et al., Citation2014). Given the differences in genetic signatures between males and females, a variety of diseases display a sexually dimorphic etiology. For example, women are much more prone to autoimmune disorders such as Lupus and rheumatoid arthritis, whereas men are more afflicted by certain cancers (e.g. hepatocellular carcinoma) and cardiovascular disease (Kaminsky et al., Citation2006). The sexual dimorphism observed in various diseases is classically driven by the primary sex hormones in each gender (testosterone in males, estrogen in females). This notion is supported by studies in which the cognate receptor for the primary sex hormone is antagonized and disease onset/progression is abated, as demonstrated by the use of the anti-estrogen molecules tamoxifen and raloxifene in the treatment of various diseases (Deroo & Korach, Citation2006). Although sex hormones clearly contribute to the sexually dimorphic phenotype, these molecules alone cannot explain the extent by which sexual dimorphism is observed, and emerging evidence suggests that certain sex differences arise from genetic makeup (XX versus XY) rather than through canonical sex-typical steroid signaling pathways per se (Smith-Bouvier et al., Citation2008).
Intriguingly, a vast array of diseases that display a sexually dimorphic etiology have an important inflammatory component to them. The inflammatory process is a balance between the initiation of inflammation and the abatement of that response. Considering that GCs are an essential physiological regulator of the anti-inflammatory response, and with sexually dimorphic diseases displaying an inflammatory component, we have speculated that GCs inherently exert their effects in a sexually dimorphic manner. To address if GCs do indeed act in a sexually dimorphic manner, Duma et al. (Citation2010) utilized the rat liver, a classical GC-responsive organ. When activation of GR was examined in response to administration of Dexamethasone, a synthetic GC, whole genome wide microarray analysis revealed a list of sexually dimorphic genes that were expanded, indicating GCs differentially regulate hepatic gene expression based on sex. Moreover, it was found that GCs act to mainly repress hepatic gene expression in males, whereas GR activation seemed to generally induce gene expression in the female rat liver. The dimorphic regulation of gene expression in response to GC treatment in males and females culminated to produce different physiological effects, with males preferentially regulating the apoptotic pathway versus females regulating the IL-6 signaling pathway (Duma et al., Citation2010). Functionally, GCs do not work as potently as anti-inflammatory molecules in females versus males in response to a global inflammatory challenge. This was demonstrated by injecting male and female Sprague Dawley rats with a lethal dose of lipopolysaccharide (LPS) and co-administering Dexamethasone. Male rats could be rescued from the global inflammatory challenge by several different doses of Dexamethasone, whereas female rats only survived when administered the highest dose of Dexamethasone (Duma et al., Citation2010). In female rats, estrogens appeared to partially play a role in inhibiting the anti-inflammatory effects of Dexamethasone since ovariectomy allowed for the rescue by lower doses of GCs, however, still not to the same efficiency as in males (Duma et al., Citation2010). Interestingly, several GC sensitive pathways that are regulated in a sexually dimorphic manner utilize Hepatic Nuclear Factor 4alpha (HNF4α) as an upstream regulator, which possibly could underlie the basis by which males and females respond to GCs differently in the liver (Duma et al., Citation2010). Besides HNF4α, there are several other transcription factors and co-activators that are enriched in males versus females and vice versa (e.g. Med12, HDAC4, STAT3).
GR/ER crosstalk as a mechanism for sexually dimorphic actions of GCs
Several transcription factors show sexual dimorphism in their expression. One of these transcription factors is the estrogen receptor (ER), another member of the nuclear receptor superfamily which acts as a transcription factor in response to ligand binding. In general, the expression of ER is much more abundant in females versus males and is enriched in organs of the female reproductive tract, though sex differences in ER expression are not uniform across all sites in the CNS. While this receptor responds to sex hormones, it is also expressed in tissues outside the reproductive tract such as liver, bone and brain, indicating a much broader physiological role than just promoting reproduction (Dupont & Gribnau, Citation2013).
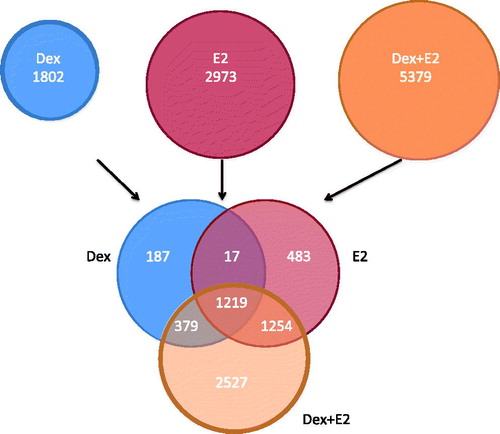
With ER being so highly expressed in females, and given the preponderance of inflammatory diseases in females, it raises the possibility that ER could possibly have antagonistic effects on the anti-inflammatory effects of GR. In fact, it has been previously shown that GCs can interfere with a number of genes regulated by estradiol, indicating at least in part, antagonism exists between these signaling pathways (Rhen et al., Citation2003). These findings were further extended by Whirledge et al. (Citation2013) to show that antagonism is bidirectional, with estradiol having inhibitory effects on GR-mediated gene regulation. In vitro studies revealed GC induction of the classical GC-induced gene, GC-induced leucine zipper (GILZ), is antagonized by the addition of estradiol – a transcriptional event dependent on ERα (Whirledge & Cidlowski, Citation2013). This antagonism is of particular interest as GILZ induction is thought to be a primary mediator of the anti-inflammatory actions of GCs. Using whole genome microarray analysis, GR/ER crosstalk has been shown to be much more extensive than just regulating the GILZ gene. While GCs and estradiol regulate unique genes when administered alone, a common set of genes appears to be co-regulated by both hormones (). Very few of these common genes are antagonistically regulated, indicating a partial overlap in function of these two hormones in human uterine epithelial cells (Whirledge et al., Citation2013). Intriguingly, when Dexamethasone and estradiol were co-administered, a large set of unique genes began to be regulated (Whirledge et al., Citation2013). There are several possibilities for these unique co-regulated genes: (1) GR/ER are only able to induce or inhibit transcription of these genes whenever both receptors are activated simultaneously or (2) there is a dynamic change in the kinetics of regulation of these genes. These unique genes could possibly be GR or ER target genes that are regulated earlier in the presence of both hormones (versus one hormone) or a gene whose transcription is extended by the presence of both hormones. Regardless of the mechanism by which these unique gene sets are regulated by co-administration of both hormones, this cohort presents previously uncharacterized targets and perhaps is an underlying basis by which GCs behave differently between the genders. While factors specific to females that antagonize GR action are very attractive possibilities to explain the sexually dimorphic actions of GCs, factors inherent to males, such as androgens, which may promote GC activity cannot be ruled out and are an area of active investigation.
Figure 1. GC and estradiol regulated genes in human uterine epithelial cells. Venn diagram of significantly regulated genes by Dexamethasone (Dex), estradiol (E2) and the combination of Dex + E2 from microarray analysis performed in human uterine epithelial cells. Source: Figure adapted from Whirledge et al., (PMID 23843231) with permission.
In conclusion, due to the high efficacy of synthetic GCs in the treatment of various inflammatory disorders and hematological cancers, pharmaceutical corticosteroids such as prednisone have become one of the most widely prescribed drugs on the market. As synthetic GCs are used so widely in the clinic, and sex is not typically a factor taken into account when prescribed, it is important to understand the natural endogenous factors that affect GC sensitivity. With work highlighting the antagonistic relationship between GR and ER and the existing clinical use of anti-estrogens, perhaps antagonizing ER with low doses of anti-estrogens will lead to increased GC sensitivity in females being treated for disorders in which synthetic GCs are routinely prescribed. Thus, sex and/or gender of the subject is a critical variable that needs to be considered for the development of personalized pharmacotherapeutic interventions when synthetic GCs are prescribed.
Bone marrow-derived myeloid cells, induced by repeated social defeat, contribute to neuroimmune responses and anxiety-like behavior
In the previous section, we provide a brief overview of how the stress- and sex-specific hormonal responses impact inflammatory gene regulation. Turning our attention back to neuroimmune mechanisms of stress, we will now discuss a well-established, highly tractable model system [repeated social defeat (RSD) in mice] that illustrates the complex interactions between peripheral immune function, stressful experiences and their impact on neuroimmune function. Specifically, the goal of this research is to examine whether stress-induced alterations in peripheral innate immunity affect central activation of microglia and development of anxiety-like behavior. RSD, a form of social stress, results in the release of myeloid cells (MCs) from the bone marrow in response to SNS activation (Engler et al., Citation2004; Hanke et al., Citation2012). These MCs are different from those released in homeostatic hematopoiesis in that they are primed and GC resistant, i.e. they are insensitive to GC-induced apoptosis (Avitsur et al., Citation2001; Engler et al., Citation2005). It appears that social stress up-regulates inflammatory gene expression in MCs via threat-related neural and endocrine signals. These signals induce changes in gene transcription, affect release of MCs into circulation, and facilitate trafficking to the brain (Wohleb et al., Citation2013). The trafficking of MCs to the brain in response to RSD occurs in the absence of tissue damage associated with pathological challenge such as traumatic brain injury, infection, stroke or autoimmune disease, and it occurs in a brain region specific manner (Wohleb et al., Citation2011, Citation2013). MCs traffic to regions associated with fear/anxiety, and the accumulation of MCs correlates with the development of prolonged anxiety-like behavior that persists for 7–14 days post stressor cessation.
In the RSD model, C57BL/6 mice are subjected to aggressive male intruder mice that are introduced into their cages for 2 hr on either one, three or six consecutive nights. During each cycle, submissive behaviors (e.g. standing upright, fleeing and crouching) are demonstrated by the resident mice, whereas aggressive behaviors (e.g. back biting and tail rattling) are demonstrated by the intruder mice. After each 2 h exposure session, the intruder mice are removed and the residents are left undisturbed until the next day when the paradigm is repeated (Avitsur et al., Citation2001). Although the potential role of injury in these neuroimmune consequences of stress cannot be totally ruled out, prior studies have failed to establish a clear association between injury in social stress models and these outcomes (e.g. Hodes et al., Citation2014; Wohleb et al., Citation2011), and social stress continues to be a well-utilized stress model in the rodent literature due to its high ethological validity (Hueston et al., Citation2011). Previous work has demonstrated that RSD induces a profound fight-or-flight response. Consistent with this, brain regions associated with fear and anxiety demonstrated neuronal activation during stressor exposure, as evidenced by increased expression of the neuronal activation marker Fos. Moreover, it was found that stress-induced anxiety-like behavior and Fos activation can be prevented by propranolol pretreatment (Hanke et al., Citation2012; Wohleb et al., Citation2011).
Social stress impacts peripheral physiologic responses as the host responds to new environmental demands caused by agonistic interactions and repeated defeat (Bailey et al., Citation2006; Engler et al., Citation2005; Kinsey et al., Citation2007). The SNS contributes to the stress response through the release of catecholamines by sympathetic nerves in lymphoid tissue; catecholamines then mediate their effects on immune cells through G-protein coupled adrenergic receptors. RSD is associated with splenomegaly and altered hematopoiesis, both of which can be blocked by β-adrenergic receptor antagonism (Engler et al., Citation2004; Powell et al., Citation2009, Citation2013). Studies from our laboratory have shown that the release of NE in the bone marrow resulted in the development of a microenvironment that supported a significant shift in hematopoiesis, resulting in enhanced myelopoiesis and the production of MCs; these cells were primed and GC resistant (Avitsur et al., Citation2003; Powell et al., Citation2013). Priming resulted in enhanced expression of Toll-like Receptors (TLRs), co-stimulatory molecules (CD80 and CD86), and increased expression of CD11b. In addition, ligation of the TLRs resulted in over-expression of proinflammatory genes (Avitsur et al., Citation2003). As mentioned previously, RSD alters hematopoiesis with a bias towards myelopoiesis; consequently primed, GC-resistant MCs are released into circulation and ultimately traffic to brain.
RSD increases the proinflammatory profile of peripheral MCs, however, the degree to which RSD affects the inflammatory profile of brain CD11b + microglia is unknown. To examine the effect of RSD on microglial activation, a series of experiments were performed on gradient enriched brain CD11b + cells (containing both CD11b + microglia and CD11b + MCs). Following 6 days of RSD, qPCR was used to study gene expression in the enriched microglia/MCs population. RSD-exposed mice showed elevated levels of gene expression for TNF-α, IL-1β, IL-6, CD14 and chemokine ligand 2 (CCL2; also known as MCP-1) (Wohleb et al., Citation2011). Ex vivo experiments using enriched cultures of CD11b + cells from the brain were stimulated with LPS for 18 h and a significant increase was seen in the production of IL-6, TNF-α and MCP-1.
Additional experiments, in which mice were given an injection of a low dose of LPS, revealed that the CD11b + cells from the brain were sensitized and had enhanced expression of genes coding for proinflammatory cytokines (IL-1β, TNF-α) and iNOS (Wohleb et al., Citation2012). Thus, both ex vivo and in vivo stimulation of brain CD11b + cells demonstrated an enhanced capacity to express genes associated with inflammation. Interestingly, ongoing studies at this time have demonstrated that the enriched brain CD11b + population is not composed entirely of microglia. Using CD45 as a biomarker, we were able to distinguish between brain CD11b + populations as microglia (CD45 + low) and brain MCs/macrophages (CD45 + high) by FACS analysis. Results from these studies have determined that 3 days of exposure to RSD was necessary to produce a significant increase in the CD45 + high population in the brain, suggesting that repeated exposure to social stress was necessary to stimulate hematopoiesis and generate primed CD11b + /CD45 + high MCs from the bone marrow (Wohleb et al., Citation2013).
Next, we asked whether peripheral MCs are recruited to the brain in response to RSD, and if so, whether MC trafficking is localized to discrete brain regions in response to RSD. To examine this question, we developed a bone marrow chimera in which GFP + (green flourescent protein; GFP) bone marrow cells were transplanted into a recipient that was conditioned using the drug busulfan to partially ablate the bone marrow. Chimeras on average had about 50% GFP + circulating MCs. In this chimera, all GFP cells were derived exclusively from the bone marrow and demonstrated greater influx of MCs into the hippocampus based on the number of cycles of RSD to which the mice were exposed (Wohleb et al., Citation2013). In fact, after six cycles of RSD the number of GFP − CD45 + cells increased approximately threefold. Importantly, these cells were found in perivascular spaces as well as deeper into the brain parenchyma. Furthermore, trafficking of the GFP + MCs was specific to brain regions associated with the response to stress (e.g. in the limbic system). Increased numbers of GFP + MCs were found in the prefrontal cortex, lateral septum (LS), amygdala and hippocampus (CA3 and dentate gyrus; Wohleb et al., Citation2013). Subsequently, we asked whether stress-induced MC recruitment to the brain was dependent on chemokine receptor expression, or simply a matter of a leaky vascular or BBB in response to RSD. In control chimeras not exposed to RSD, neither the brain vasculature nor the BBB was found to be leaky (Wohleb et al., Citation2013), suggesting that passive changes in the blood vasculature could not account for the passage of GFP + cells into the CNS after RSD. At about this same time, studies (Wohleb et al., Citation2013) examining two chemokine receptors associated with the MC trafficking – C–C chemokine receptor 2 (CCR2) and CX3C chemokine receptor 1 (CX3CR1; also known as fractalkine receptor) – demonstrated that RSD promoted prolonged anxiety in wild type mice, but not in mice deficient in the expression of these chemokine receptors. Importantly, these chemokine receptor KO mice did not traffic MPCs to brain, nor did they develop anxiety-like behavior in response to RSD. Thus, the expression of chemokines in response to RSD appears to be essential for the trafficking of MCs into the CNS as well as the heightened anxiety-like behavior produced by RSD exposure.
It appears that resident microglia and brain macrophages (derived from circulating MCs) contribute to RSD-induced, prolonged anxiety. This behavioral response requires proinflammatory cytokine gene expression (TNF-α, IL-1β) and chemokine gene expression (CCR2 and CX3CR1). Further experiments are required to support the concept of region-specific trafficking of MCs. The next logical step, therefore, is to demonstrate the development of a reactive endothelium to facilitate cell adhesion to the vasculature in regions associated with extravasation of MCs. This has raised several critical questions, including: What happens after cessation of RSD? Are these animals sensitized by RSD? Is there evidence of macrophage/microglia priming weeks after RSD? Indeed, we have observed that by 24 days after RSD, the number of bone marrow-derived macrophages in the brain was significantly reduced and anxiety-like behavior was largely resolved. However, analysis of proinflammatory gene expression in CD11b + microglia from the brain indicated that the microglia maintained a “primed” mRNA profile (enhanced expression of proinflammatory genes upon stimulation). In summary, by 24 days after RSD, behavior returned to normal, the presence of macrophages in the brain was significantly reduced, but microglia maintained a primed/sensitized state (Wohleb et al., Citation2014).
Is microglia priming relevant to a model of long-term stress sensitization? In other words, does exposure of primed mice to a sub-threshold dose of the original stressor result in an anxiety-like response that correlates with the trafficking of MCs to the brain, perhaps up to months after initial exposure to RSD? From previous studies it was known that a single exposure to RSD was not sufficient to stimulate production of MCs from bone marrow and did not cause an increase in anxiety-like behavior (Avitsur et al., Citation2001). However, a single exposure to RSD re-established anxiety in stress-sensitized mice, and stimulated the re-appearance of GFP + MCs in the brain. The source of the GFP + MCs in the brain was actually found to be a cellular depot of primed MCs located in the spleen, which developed in response to the initial exposures to RSD. Consistent with this, removal of the spleen (splenectomy) in stress-sensitized mice abrogated anxiety and prevented GFP + MCs from trafficking to the brain in response to a single exposure to RSD (Wohleb et al., Citation2014). Thus, it appears that MCs activated by the original cycles of RSD take up residence in the spleen upon stress cessation, and after several weeks without stress, these cells (now residing in the spleen) can be re-activated to transit into the CNS where, among other things, they appear to re-instate anxiety-like behavior.
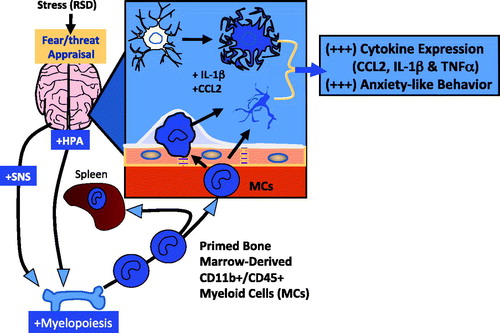
To briefly summarize, in a mouse model of RSD, prolonged anxiety-like behavior was associated with microglia activation and the trafficking of bone marrow-derived peripheral MCs to the brain. In response to the stressor, MCs were primed and GC resistant. Upon stimulation, primed MCs produced high levels of proinflammatory cytokine gene expression. These primed MCs extravasated to brain regions associated with fear and anxiety during stress, with trafficking to the brain requiring the expression of two chemokine receptors, CCR2 and CX3CR1. The role of peripheral bone marrow-derived MCs in the development and prolongation of anxiety-like behavior is a unique feature of this model of RSD and is summarized in . In addition to providing an excellent model for better understanding the complex interactions between neural and immune responses to stress, the RSD model illustrates the dynamic physiology underlying neuroimmune consequences of stress and their implications for understanding affective consequences of repeated stressor exposure. Considering that RSD exposure led to GC resistance, these findings also underscore the importance of stress history as a critical determinant of drug responsiveness, particularly in cases where inflammatory pathways are being targeted for pharmacotherapeutic intervention.
Figure 2. Microglia and CD11b + /CD45 MCs contribute to social defeat-induced prolonged anxiety. The response to social defeat activates fear/threat appraisal circuitry in the brain. Activation of microglia in these areas results in the release proinflammatory cytokines including IL-1β and CCL2. In turn, these cytokine responses contribute to the development of a reactive endothelium in the regional neurovasculature. Social defeat also activates the HPA axis and SNS which stimulate the production of primed CD11 + /CD45 + MCs in the bone marrow. Release of MCs into circulation results in trafficking of these cells to the reactive neurovasculature which is followed by adhesion and diapedesis into the brain. The activation of microglia and the trafficking of primed MCs results in the development of prolonged anxiety-behavior.
Long-term consequences of early life pain on adult stress responses
The studies reviewed thus far have focused exclusively on subjects in which neuroimmune mechanisms of stress have been manipulated in adult subjects. However, a long history of evidence suggests that the impact of stress may vary as a function of the developmental epoch in which the challenge is imposed (e.g. Levine, Citation2005). Emerging evidence suggests that developmental timing of inflammatory challenges also has a profound influence on the developmental trajectory of stress responsive systems, which may in turn impact vulnerability of adult subjects to later stress-related pathologies (Bilbo & Schwarz, Citation2012). In the section that follows, we will describe a series of studies in which neonatal inflammation in a rat model substantially impacts adult stress physiology.
Premature birth, defined as birth prior to 37 weeks of gestation, occurs at alarmingly high rates worldwide. According to the World Health Organization, 16.5% of all infants are born premature, with over 500,000 preterm babies born each year in the USA alone (Martin et al., Citation2006). Preterm infants spend, on average, 20 days in the Neonatal Intensive Care Unit (NICU), where they undergo 10–18 invasive procedures each day, including heel lances, endotracheal intubation, surgery, and respiratory and gastric suctioning (Barker & Rutter, Citation1995; Simons et al., Citation2003). These procedures typically result in an inflammatory response at the site of injury, invoking pain and stress in the newborn. As premature infants are traditionally thought to be incapable of feeling pain due to an immature and underdeveloped sensory system, most of these procedures occur in the absence of preemptive and/or reactive use of anesthetic or analgesic treatment. However, recent studies using advanced imaging techniques have shown that acute noxious stimuli do indeed evoke cortical activity in preterm infants as young as 25 weeks (Bartocci et al., Citation2006; Slater et al., Citation2006). Similarly, preterm infants have robust catecholamine and stress hormone responses to noxious, but not non-noxious procedures (Anand et al., Citation1987; Grunau et al., Citation2004). Despite strong evidence that pain and stress circuitry are established and functional during late gestation, specific anesthesia is only used in 5–21% of the invasive procedures routinely performed in the NICU (Anand & Carr, Citation1989; Carbajal et al., Citation2008; Giannakoulopoulos et al., Citation1999; Meaney et al., Citation1985; Sapolsky et al., Citation1985; Simons et al., Citation2003; Walker et al., Citation1986a, Citation1986b; Walter-Nicolet et al., Citation2010).
Growing clinical and basic science data suggest that exposure to repeated tissue damaging procedures in neonates with limited analgesic therapy induces lasting changes in the brain and spinal cord, which has been shown to have profound consequences for subsequent nociceptive processing (Anand, Citation2000; Bhutta et al., Citation2001; Grunau et al., Citation2005; Lidow, Citation2002; Ren et al., Citation2004; Walker et al., Citation2003; Wang et al., Citation2004; Whitfield & Grunau, Citation2000). In addition to changes in pain sensitivity, clinical studies suggest NICU treatment has an immediate and long-term impact on the response to stress- and anxiety-provoking stimuli. Intrinsically, painful NICU procedures activate the stress response (Anand et al., Citation1987; Bouza, Citation2009; Peters et al., Citation2005; Walter-Nicolet et al., Citation2010). Moreover, preterm infants undergoing surgical procedures have significantly higher concentrations of catecholamines and GCs during and after surgery as compared with infants receiving analgesic treatment (Anand et al., Citation1987). While initially heart rate, facial reactivity and cortisol levels of preterm infants were high in response to procedural pain, these responses became significantly blunted as the number of skin breaking procedures increased (Grunau et al., Citation2001, Citation2010a). Even in early childhood, cortisol release in response to painful stimuli remained blunted in former preterm infants (Grunau et al., Citation2007, Citation2010a).
Longitudinal studies focusing on the behavioral impact of NICU treatment found that, by adolescence, infants born weighing <1500 g (very low birth weight; VLBW) were at least 28% more likely to suffer from neuropsychiatric disorders including issues with anxiety, depression and attention deficit hyperactivity (Botting et al., Citation1997; Levy-Shiff et al., Citation1994). Furthermore, 20-year-olds born at VLBW self-reported significantly more clinical behaviors of internalization, such as anxiety, depression, social withdrawal and inattention relative to normal birth weight controls, as did their parents (Aarnoudse-Moens et al., Citation2009; Hack et al., Citation2004; Hayes & Sharif, Citation2009; Sullivan et al., Citation2012). Similar to clinical findings of long-term changes in pain sensitivity, these studies suggest that early life pain experienced in the NICU is associated with putatively permanent changes in stress physiology and behavior.
Results from a rodent model of early life pain mirror what has been reported clinically. In this model, newborn rats (postnatal day 1; developmentally equivalent to a 24–36 week gestation infant) receive an intraplantar injection of the inflammatory agent carrageenan (CGN). Intraplantar CGN results in a local inflammatory response that lasts between 24 and 72 h and approximates the inflammatory period of common skin-breaking procedures in the NICU (LaPrairie et al., Citation2008; LaPrairie & Murphy, Citation2007, Citation2009; Peters et al., Citation2005; Simons et al., Citation2003). Importantly, this model of early life injury did not alter maternal behavior directed toward the pup (LaPrairie & Murphy, Citation2007), nor does it affect normal development in terms of weight gain and the onset of puberty (LaPrairie & Murphy, Citation2007). We have also reported that early life inflammatory pain resulted in decreased responsiveness to acute stress- and anxiety-provoking stimuli in adulthood (postnatal days 60–90) (Victoria et al., Citation2013b,Citationc, Citation2014). For example, rats that experienced unresolved pain on the day of birth displayed five- to sixfold increases in their latency to exhibit immobility in the forced swim test, spent a significantly greater amount of time in the inner area of the open field, and demonstrated dampened corticosterone levels following acute restraint (Victoria et al., Citation2013c). These changes in responsiveness to acute stress and anxiety were accompanied by significant increases in pro-enkephalin mRNA expression within the periaqueductal gray (PAG), central amygdala and LS. Indeed, changes in endogenous opioid tone likely contributed to the observed hypo-responsive profile, as administration of the broad-spectrum opioid antagonist naloxone reversed the effects of early life pain (Victoria et al., Citation2013c).
Collectively, our results demonstrate that neonatal injury decreases adult responses to acute stress- and anxiety-provoking stimuli (hypo-responsive state). These findings are consistent with clinical studies reporting blunted responses to stress, high levels of emotional internalization, altered ability to externalize, reduced ability to adapt, and cognitive inflexibility for former preterm infants (Badanes et al., Citation2011; Botting et al., Citation1997; Grunau et al., Citation2001, Citation2007, Citation2010b; Levy-Shiff et al., Citation1994). Clinically, decreases in reactivity to anxiogenic, stressful or rewarding stimuli are hallmarks of affective dysfunction (Krishnan & Nestler, Citation2008). Therefore, it is not surprising that infants who have experienced early life pain and/or trauma are at a higher risk for developing affective disorders later in life.
Early life pain is also associated with changes in receptors that regulate stress activation and recovery. Using competitive receptor autoradiography, neonatally injured adults had significantly decreased binding of the corticotrophin receptor type 1 (CRFR1) in the basolateral amygdala and PAG as compared to controls. In contrast, CRFR2 binding was significantly increased in the LS and cortical amygdala of neonatally injured adults relative to controls. As activation of the stress response is associated with CRFR1 (Vale et al., Citation1981), whereas CRFR2 promotes return to homeostasis following a perturbation (Bale et al., Citation2000; Coste et al., Citation2000), these findings are consistent with data demonstrating that early life pain blunts adult behavioral sensitivity to stress-, anxiety- and pain-provoking stimuli (LaPrairie & Murphy, Citation2007, Citation2009; Victoria et al., Citation2013a).
In addition to changes in CRF receptor expression, GR mRNA and protein were increased in the PVN but decreased in the hippocampus of neonatally injured adults in comparison to controls. Decreased GR expression in the hippocampus has been associated with psychiatric conditions, including mood and anxiety disorders. Indeed, early life adversity is one of the most prominent environmental factors associated with an increased risk of developing mood and anxiety disorders (Gross & Hen, Citation2004; Heim & Nemeroff, Citation2001). A growing body of evidence suggests that early environmental factors can have long-term behavioral and neuroendocrine consequences in rodents (Francis et al., Citation2003; Liu et al., Citation1997, Citation2000; Meaney, Citation2001). In rats, paradigms designed to model abuse and neglect have repeatedly shown that early traumatic experience permanently decreases GR expression in the hippocampus. Humans that experience abuse or maltreatment also have decreased GRs in the hippocampus, and this has been proposed to underlie disrupted HPA reactivity to stressors and increased susceptibility to psychiatric disorders (McGowan et al., Citation2009). In rodents, early adverse experiences have been shown to lead to permanent epigenetic chemical modifications of the GR promoter and we hypothesize that neonatal pain may have similar life-long molecular and psychopathological effects as abuse and maltreatment.
Collectively, these data suggest that a single neonatal injury alters receptor systems and neural circuits that contribute to activation of the stress axis, as well as neuroendocrine recovery from stress. Thus, these alterations likely contribute to the blunted behavioral responses previously observed in response to stress- and anxiety-provoking stimuli. Such long-term changes are likely the result of unrelieved pain and its associated distress, given evidence that administration of morphine at the time of injury mitigated the observed hormonal and behavioral responses to anxiety- and stress-provoking stimuli (Bhutta et al., Citation2001; LaPrairie et al., Citation2008; Victoria et al., Citation2014). Importantly, these results indicate a need for specific and appropriate analgesic regimes for managing early life pain. Remarkably, less than 35% of NICU patients receive therapy for the multitude of invasive, painful procedures experienced each day in the NICU (Barker & Rutter, Citation1995; Carbajal et al., Citation2008; Simons et al., Citation2003). Results from a recent study demonstrating that former preterm infants who received morphine for pain management in the NICU had improved executive functioning and reduced problems with externalization at 8–9 years of age (relative to those that received placebo) may help mitigate this practice (de Graaf et al., Citation2013).
When considering these findings, two key issues on the potential role of inflammation require discussion. First, the neonatal pain stimulus utilized in the studies described above was CGN, which is a widely used rodent model of inflammatory pain. This raises the intriguing question of whether the apparent life-long alterations in stress reactivity observed in CGN-injected rats resulted from the release of inflammation-dependent signaling factors (cytokines, chemokines, prostaglandins, etc.). Indeed, prior studies clearly indicate a strong role for glia (microglia, astrocytes, etc.) in the propagation of pain signals, indicating a prominent role for cytokines in the development of pathological pain processes and other sensitization phenomena (Grace et al., Citation2014a; Watkins et al., Citation2001). Consistent with this, use of viral vectors to deliver anti-inflammatory cytokines/chemokines seems to effectively reverse long-term changes in pain sensitivity evoked by inflammation (Milligan et al., Citation2005; Sloane et al., Citation2009). Looking beyond pain signals, one must also consider recent evidence showing that opioid analgesics such as morphine interact with Toll-Like Receptor 4 (TLR4), a pathogen-recognition receptor whose activation spurs the expression of cytokines (Grace et al., Citation2014b; Hutchinson et al., Citation2012), suggesting that either endogenous opioids (released in response to noxious stimuli) or exogenous opiates (to control pain) might exert certain effects through inflammatory signaling pathways downstream of TLR4 activation. As such, there appear to be a multitude of mechanisms by which noxious (or antigenic) stimuli intersect with neuroimmune signaling pathways during early development and have the ability to produce profound re-architecture of the stress axis. Future studies seeking to further dissect the developmental vulnerabilities and neuroimmune mechanisms of these changes will significantly contribute to the development of novel therapeutics to target stress-related pathologies that may relate to early life experience.
Summary and conclusions
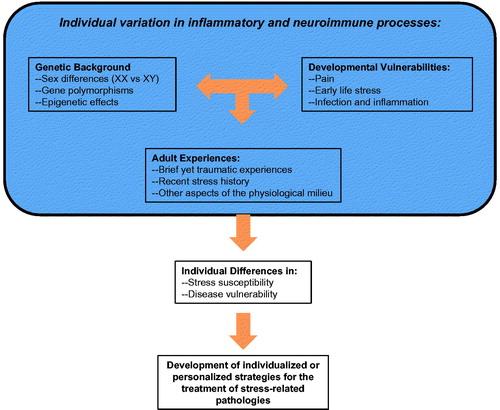
Our goal in preparation of this article was to highlight the importance of neuroimmune consequences of stress as critical mediators of later stress sensitivity and ultimately stress-related pathologies. We utilized an exemplar approach with review of relevant literature to forge a bridge between classic aspects of stress physiology (SNS and HPA axis hormones) and neuroimmune function, with significant discussion of individual subject characteristics (sex, stress history, developmental experience) that may determine the neuroimmune response profile observed (depicted in ). In doing so, several implications for the future arose. First, steady-state levels of neuroinflammation appear to escalate in response to distinct stress challenges. Importantly, the nature of the neuroimmune response evoked by stress depends on key features of the individual, including genetic differences (sex and other genomic differences) and/or hormonal status of the subject (Arakawa et al., Citation2014; Duma et al., Citation2010; Hodes et al., Citation2014). Experiential factors, such as social experiences, appear to be particularly important drivers of neuroimmune differences across a wide range of species. For instance, the presence of dominance hierarchies in pair-housed male rats is predictive of ambient IL-1 concentration in brain (Barnum et al., Citation2008); RSD in male mice altered chemokine-mediated recruitment of MCs to the CNS (Wohleb et al., Citation2013); and maternal separation of guinea pig pups produced a constellation of sickness-like behaviors that are ameliorated by drugs with anti-inflammatory properties (Hennessy et al., Citation2015). Finally, the identification of specific developmental epochs during which individuals may be susceptible (vulnerable) to stress-, pain- or inflammation-dependent changes in later stress reactivity also emerged as key individual drivers of enhanced stress reactivity later in life. Together, these individual differences provide strategic starting blocks for the development of personalized or individualized pharmacotherapeutic approaches that can be used to effectively treat stress-related pathology.
Figure 3. Summary of key factors to be considered in the development of individualized treatment strategies for stress-related disorders.
Declaration of interest
This review summarizes a series of talks given at the Neurobiology of Stress Workshop in Cincinnati, OH from 17–20 June 2014. Dr. Terrence Deak is currently supported by NIH grants P50AA017823 and RO1AG043467, and a contract with Janssen Pharmaceuticals, Inc. Dr. Matt Quinn and Dr. John A. Cidlowski are currently funded by the National Institutes of Environmental Health/National Institutes of Health. Research presented by Dr. Nicole C. Victoria and Dr. Anne Z. Murphy was funded by DA16272. Dr. John F. Sheridan is currently funded by NIH grants RO1MH093473 and RO1MH097243. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the above stated funding agencies. The authors have no conflicts of interest to declare.
References
- Aarnoudse-Moens CS, Weisglas-Kuperus N, van Goudoever JB, Oosterlaan J. (2009). Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics 124:717–28
- American Psychological Association. (2012). Report on Stress in America. Washington, DC: APA
- Anand KJ. (2000). Pain, plasticity, and premature birth: a prescription for permanent suffering? Nat Med 6:971–3
- Anand KJ, Carr DB. (1989). The neuroanatomy, neurophysiology, and neurochemistry of pain, stress, and analgesia in newborns and children. Pediatr Clin North Am 36:795–822
- Anand KJ, Sippell WG, Aynsley-Green A. (1987). Randomised trial of fentanyl anaesthesia in preterm babies undergoing surgery: effects on the stress response. Lancet 1:243–8
- Anisman H. (2009). Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci 34:4–20
- Arakawa K, Arakawa H, Hueston CM, Deak T. (2014). Effects of the estrous cycle and ovarian hormones on central expression of interleukin-1 evoked by stress in female rats. Neuroendocrinology 100:162--77
- Arakawa H, Blandino Jr P, Deak T. (2009). Central infusion of interleukin-1 receptor antagonist blocks the reduction in social behavior produced by prior stressor exposure. Physiol Behav 98:139–46
- Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H, Sheridan JF. (2003). Expression of glucocorticoid resistance following social stress requires a second signal. J Leukocyte Biol 74:507–13
- Avitsur R, Stark JL, Sheridan JF. (2001). Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav 39:247–57
- Badanes LS, Watamura SE, Hankin BL. (2011). Hypocortisolism as a potential marker of allostatic load in children: associations with family risk and internalizing disorders. Dev Psychopathol 23:881–96
- Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. (2011). Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun 25:397–407
- Bailey MT, Engler H, Sheridan JF. (2006). Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J Neuroimmunol 171:29–37
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, et al. (2000). Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet 24:410–14
- Barker DP, Rutter N. (1995). Exposure to invasive procedures in neonatal intensive care unit admissions. Arch Dis Child Fetal Neonatal Ed 72:F47–8
- Barnum CJ, Blandino Jr P, Deak T. (2008). Social status modulates basal IL-1 concentrations in the hypothalamus of pair-housed rats and influences certain features of stress reactivity. Brain Behav Immun 22:517–27
- Bartocci M, Bergqvist LL, Lagercrantz H, Anand KJ. (2006). Pain activates cortical areas in the preterm newborn brain. Pain 122:109–17
- Bhutta AT, Rovnaghi C, Simpson PM, Gossett JM, Scalzo FM, Anand KJ. (2001). Interactions of inflammatory pain and morphine in infant rats: long-term behavioral effects. Physiol Behav 73:51–8
- Bilbo SD, Schwarz JM. (2012). The immune system and developmental programming of brain and behavior. Front Neuroendocrinol 33:267–86
- Black PH, Garbutt LD. (2002). Stress, inflammation and cardiovascular disease. J Psychosom Res 52:1–23
- Blandino P Jr, Barnum CJ, Deak T. (2006). The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol 173:87–95
- Blandino P Jr, Barnum CJ, Solomon LG, Larish Y, Lankow BS, Deak T. (2009). Gene expression changes in the hypothalamus provide evidence for regionally-selective changes in IL-1 and microglial markers after acute stress. Brain Behav Immun 23:958–68
- Blandino P Jr, Hueston CM, Barnum CJ, Bishop C, Deak T. (2013). The impact of ventral noradrenergic bundle lesions on increased IL-1 in the PVN and hormonal responses to stress in male sprague dawley rats. Endocrinology 154:2489–500
- Botting N, Powls A, Cooke RW, Marlow N. (1997). Attention deficit hyperactivity disorders and other psychiatric outcomes in very low birthweight children at 12 years. J Child Psychol Psychiatry 38:931–41
- Bouza H. (2009). The impact of pain in the immature brain. J Matern Fetal Neonatal Med 22:722–32
- Carbajal R, Rousset A, Danan C, Coquery S, Nolent P, Ducrocq S, Saizou C, et al. (2008). Epidemiology and treatment of painful procedures in neonates in intensive care units. JAMA 300:60–70
- Catanzaro JM, Hueston CM, Deak MM, Deak T. (2014). The impact of the P2X7 receptor antagonist A-804598 on neuroimmune and behavioral consequences of stress. Behav Pharmacol 25:582–98
- Cohen BE, Panguluri P, Na B, Whooley MA. (2010). Psychological risk factors and the metabolic syndrome in patients with coronary heart disease: findings from the Heart and Soul Study. Psychiatry Res 175:133–7
- Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, et al. (2000). Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet 24:403–9
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
- de Graaf J, van Lingen RA, Valkenburg AJ, Weisglas-Kuperus N, Groot Jebbink L, Wijnberg-Williams B, Anand KJ, et al. (2013). Does neonatal morphine use affect neuropsychological outcomes at 8 to 9 years of age? Pain 154:449–58
- Deak T. (2007). From classic aspects of the stress response to neuroinflammation and sickness: implications for individuals and offspring of diverse species. Int J Comp Psychol 20:96–110
- Deak T. (2008). Immune cells and cytokine circuits: toward a working model for understanding direct immune-to-adrenal communication pathways. Endocrinology 149:1433–5
- Deak T, Bellamy C, D'Agostino LG. (2003). Exposure to forced swim stress does not alter central production of IL-1. Brain Res 972:53–63
- Deak T, Bordner KA, McElderry NK, Barnum CJ, Blandino P Jr, Deak MM, Tammariello SP. (2005). Stress-induced increases in hypothalamic IL-1: a systematic analysis of multiple stressor paradigms. Brain Res Bull 64:541–56
- Deak T, Nguyen KT, Fleshner M, Watkins LR, Maier SF. (1999). Acute stress may facilitate recovery from a subcutaneous bacterial challenge. Neuroimmunomodulation 6:344–54
- Deroo BJ, Korach KS. (2006). Estrogen receptors and human disease. J Clin Invest 116:561–70
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. (1995). Effects of stress on immune cell distribution. Dynamics and hormonal mechanisms. J Immunol 154:5511–27
- Duma D, Collins JB, Chou JW, Cidlowski JA. (2010). Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal 3:ra74
- Dupont C, Gribnau J. (2013). Different flavors of X-chromosome inactivation in mammals. Curr Opin Cell Biol 25:314–21
- Elenkov IJ, Chrousos GP. (2002). Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann NY Acad Sci 966:290–303
- Engler H, Bailey MT, Engler A, Sheridan JF. (2004). Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol 148:106–15
- Engler H, Engler A, Bailey MT, Sheridan JF. (2005). Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. J Neuroimmunol 163:110–19
- Etgen AM, Barfield RJ. (1986). Antagonism of female sexual behavior with intracerebral implants of antiprogestin RU 38486: correlation with binding to neural progestin receptors. Endocrinology 119:1610–17
- Francis DD, Szegda K, Campbell G, Martin WD, Insel TR. (2003). Epigenetic sources of behavioral differences in mice. Nat Neurosci 6:445–6
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. (2007). Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21:47–59
- Frank MG, Barrientos RM, Watkins LR, Maier SF. (2010). Aging sensitizes rapidly isolated hippocampal microglia to LPS ex vivo. J Neuroimmunol 226:181–4
- Frank MG, Hershman SA, Weber MD, Watkins LR, Maier SF. (2014). Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 40:191–200
- Frank MG, Watkins LR, Maier SF. (2013). Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav Immun 33:1–6
- Garcia-Bueno B, Madrigal JL, Perez-Nievas BG, Leza JC. (2008). Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology 149:1969–78
- Giannakoulopoulos X, Teixeira J, Fisk N, Glover V. (1999). Human fetal and maternal noradrenaline responses to invasive procedures. Pediatr Res 45:494–9
- Goshen I, Yirmiya R, Iverfeldt K, Weidenfeld J. (2003). The role of endogenous interleukin-1 in stress-induced adrenal activation and adrenalectomy-induced adrenocorticotropic hormone hypersecretion. Endocrinology 144:4453–8
- Grace PM, Hutchinson MR, Maier SF, Watkins LR. (2014a). Pathological pain and the neuroimmune interface. Nat Rev Immunol 14:217–31
- Grace PM, Ramos KM, Rodgers KM, Wang X, Hutchinson MR, Lewis MT, Morgan KN, et al. (2014b). Activation of adult rat CNS endothelial cells by opioid-induced toll-like receptor 4 (TLR4) signaling induces proinflammatory, biochemical, morphological, and behavioral sequelae. Neuroscience 280:299–317
- Gross C, Hen R. (2004). The developmental origins of anxiety. Nat Rev Neurosci 5:545–52
- Grunau RE, Haley DW, Whitfield MF, Weinberg J, Yu W, Thiessen P. (2007). Altered basal cortisol levels at 3, 6, 8 and 18 months in infants born at extremely low gestational age. J Pediatr 150:151–6
- Grunau RE, Holsti L, Haley DW, Oberlander T, Weinberg J, Solimano A, Whitfield MF, et al. (2005). Neonatal procedural pain exposure predicts lower cortisol and behavioral reactivity in preterm infants in the NICU. Pain 113:293–300
- Grunau RE, Oberlander TF, Whitfield MF, Fitzgerald C, Lee SK. (2001). Demographic and therapeutic determinants of pain reactivity in very low birth weight neonates at 32 Weeks' postconceptional Age. Pediatrics 107:105–12
- Grunau RE, Tu MT, Whitfield MF, Oberlander TF, Weinberg J, Yu W, Thiessen P, et al. (2010a). Cortisol, behavior, and heart rate reactivity to immunization pain at 4 months corrected age in infants born very preterm. Clin J Pain 26:698–704
- Grunau RE, Weinberg J, Whitfield MF. (2004). Neonatal procedural pain and preterm infant cortisol response to novelty at 8 months. Pediatrics 114:e77–84
- Grunau BE, Wiens MO, Brubacher JR. (2010b). Dantrolene in the treatment of MDMA-related hyperpyrexia: a systematic review. CJEM 12:435–42
- Hack M, Youngstrom EA, Cartar L, Schluchter M, Taylor HG, Flannery D, Klein N, Borawski E. (2004). Behavioral outcomes and evidence of psychopathology among very low birth weight infants at age 20 years. Pediatrics 114:932–40
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF. (2012). Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26:1150–9
- Hayes B, Sharif F. (2009). Behavioural and emotional outcome of very low birth weight infants – literature review. J Matern Fetal Neona 22:849–56
- Heim C, Nemeroff CB. (2001). The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry 49:1023–39
- Hennessy MB, Paik KD, Caraway JD, Schiml PA, Deak T. (2011). Proinflammatory activity and the sensitization of depressive-like behavior during maternal separation. Behav Neurosci 125:426–33
- Hennessy MB, Stafford NP, Yusko-Osborne B, Schiml PA, Xanthos ED, Deak T. (2015). Naproxen attenuates sensitization of depressive-like behavior and fever during maternal separation. Physiol Behav 139:34–40
- Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, Rebusi N, et al. (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA 111:16136–41
- Hueston CM, Barnum CJ, Eberle JA, Ferraioli FJ, Buck HM, Deak T. (2011). Stress-dependent changes in neuroinflammatory markers observed after common laboratory stressors are not seen following acute social defeat of the Sprague Dawley rat. Physiol Behav 104:187–98
- Hueston CM, Deak T. (2014). The inflamed axis: the interaction between stress, hormones, and the expression of inflammatory-related genes within key structures comprising the hypothalamic–pituitary–adrenal axis. Physiol Behav 124:77–91
- Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, et al. (2012). Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci 32:11187–200
- Jeong YH, Park CH, Yoo J, Shin KY, Ahn SM, Kim HS, Lee SH, et al. (2006). Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. FASEB J 20:729–31
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. (2005). Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 135:1295–307
- Johnson JD, Fleshner M. (2006). Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J Leukocyte Biol 79:425–34
- Johnson JD, O'Connor KA, Deak T, Spencer RL, Watkins LR, Maier SF. (2002a). Prior stressor exposure primes the HPA axis. Psychoneuroendocrinology 27:353–65
- Johnson JD, O'Connor KA, Deak T, Stark M, Watkins LR, Maier SF. (2002b). Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun 16:461–76
- Kaminsky Z, Wang SC, Petronis A. (2006). Complex disease, gender and epigenetics. Ann Med 38:530–44
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. (2007). Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun 21:458–66
- Krishnan V, Nestler EJ. (2008). The molecular neurobiology of depression. Nature 455:894–902
- LaPrairie JL, Johns ME, Murphy AZ. (2008). Preemptive morphine analgesia attenuates the long-term consequences of neonatal inflammation in male and female rats. Pediatr Res 64:625–30
- LaPrairie JL, Murphy AZ. (2007). Female rats are more vulnerable to the long-term consequences of neonatal inflammatory injury. Pain 132:S124–33
- LaPrairie JL, Murphy AZ. (2009). Neonatal injury alters adult pain sensitivity by increasing opioid tone in the periaqueductal gray. Front Behav Neurosci 3:31 (1--11)
- Levine S. (2005). Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology 30:939–46
- Levy-Shiff R, Einat G, Mogilner MB, Lerman M, Krikler R. (1994). Biological and environmental correlates of developmental outcome of prematurely born infants in early adolescence. J Pediatr Psychol 19:63–78
- Lidow MS. (2002). Long-term effects of neonatal pain on nociceptive systems. Pain 99:377–83
- Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. (2000). Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci 3:799–806
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, et al. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic–pituitary–adrenal responses to stress. Science 277:1659–62
- Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, Lovenberg TW, et al. (2014). Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J Pharm Exp Ther 351:628–41
- Macht M, Haupt C, Ellgring H. (2005). The perceived function of eating is changed during examination stress: a field study. Eating Behav 6:109–12
- Maes M, Fisar Z, Medina M, Scapagnini G, Nowak G, Berk M. (2012). New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates – Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 20:127–50
- Maier SF, Watkins LR. (1995). Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res 695:279–82
- Maier SF, Watkins LR. (1998). Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev 105:83–107
- Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Kirmeyer S. (2006). Births: final data for 2004. Natl Vital Stat Rep 55:1–101
- Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. (2014). Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci 34:15490–6
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 12:342–8
- Meaney MJ. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 24:1161–92
- Meaney MJ, Aitken DH, Bodnoff SR, Iny LJ, Sapolsky RM. (1985). The effects of postnatal handling on the development of the glucocorticoid receptor systems and stress recovery in the rat. Prog Neuropsychopharmacol Biol Psychiatry 9:731–4
- Miller AH. (2008). Inflammation versus glucocorticoids as purveyors of pathology during stress: have we reached the tipping point? Biol Psychiatry 64:263–5
- Milligan ED, Nguyen KT, Deak T, Hinde JL, Fleshner M, Watkins LR, Maier SF. (1998). The long term acute phase-like responses that follow acute stressor exposure are blocked by alpha-melanocyte stimulating hormone. Brain Res 810:48–58
- Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, Wieseler-Frank J, et al. (2005). Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci 21:2136--48
- Nair A, Bonneau RH. (2006). Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol 171:72–85
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. (1998). Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci 18:2239–46
- Nguyen KT, Deak T, Will MJ, Hansen MK, Hunsaker BN, Fleshner M, Watkins LR, Maier SF. (2000). Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1beta protein response to acute stress. Brain Res 859:193–201
- Oakley RH, Cidlowski JA. (2011). Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286:3177–84
- Oakley RH, Cidlowski JA. (2013). The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–44
- Pace TW, Heim CM. (2011). A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav Immun 25:6–13
- Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM. (2006). Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–3
- Peters JW, Schouw R, Anand KJ, van Dijk M, Duivenvoorden HJ, Tibboel D. (2005). Does neonatal surgery lead to increased pain sensitivity in later childhood? Pain 114:444–54
- Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, Sheridan JF. (2009). Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun 23:225–31
- Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, et al. (2013). Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA 110:16574–9
- Quinn M, Ramamoorthy S, Cidlowski JA. (2014). Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Ann NY Acad Sci 1317:1–6
- Ren K, Anseloni V, Zou SP, Wade EB, Novikova SI, Ennis M, Traub RJ, et al. (2004). Characterization of basal and re-inflammation-associated long-term alteration in pain responsivity following short-lasting neonatal local inflammatory insult. Pain 110:588–96
- Rhen T, Grissom S, Afshari C, Cidlowski JA. (2003). Dexamethasone blocks the rapid biological effects of 17beta-estradiol in the rat uterus without antagonizing its global genomic actions. FASEB J 17:1849–70
- Rupprecht R, Reul JM, Trapp T, van Steensel B, Wetzel C, Damm K, Zieglgänsberger W, Holsboer F. (1993). Progesterone receptor-mediated effects of neuroactive steroids. Neuron 11:523–30
- Sapolsky RM, Meaney MJ, McEwen BS. (1985). The development of the glucocorticoid receptor system in the rat limbic brain. III. Negative-feedback regulation. Brain Res 350:169–73
- Schmid W, Cole TJ, Blendy JA, Schutz G. (1995). Molecular genetic analysis of glucocorticoid signalling in development. J Steroid Biochem Mol Biol 53:33–5
- Simons SH, van Lingen RA, Tibboel D. (2003). Do we still hurt newborn babies? A prospectie study of procedural pain and analgesia in neonates. Arch Pediatric Adolesc Med 157:1058–64
- Slater R, Cantarella A, Gallella S, Worley A, Boyd S, Meek J, Fitzerald M. (2006). Cortical pain responses in human infants. J Neurosci 26:3662–6
- Sloane EM, Soderquist RG, Maier SF, Mahoney MJ, Watkins LR, Milligan ED. (2009). Long-term control of neuropathic pain in a non-viral gene therapy paradigm. Gene Ther 16:470–5
- Smith-Bouvier DL, Divekar AA, Sasidhar M, Du S, Tiwari-Woodruff SK, King JK, Arnold AP, et al. (2008). A role for sex chromosome complement in the female bias in autoimmune disease. J Exp Med 205:1099–108
- Sorrells SF, Sapolsky RM. (2007). An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun 21:259–72
- Stein DG. (2008). Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev 57:386–97
- Sternberg EM. (2001). Neuroendocrine regulation of autoimmune/inflammatory disease. J Endocrinol 169:429–35
- Sullivan MC, Msall ME, Miller RJ. (2012). 17-Year outcome of preterm infants with diverse neonatal morbidities: Part 1 – impact on physical, neurological, and psychological health status. J Spec Pediatr Nurs 17:226–41
- Vale W, Spiess J, Rivier C, Rivier J. (1981). Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394–7
- van der Burg B, van der Saag PT. (1996). Nuclear factor-kappa-B/steroid hormone receptor interactions as a functional basis of anti-inflammatory action of steroids in reproductive organs. Mol Human Reprod 2:433–8
- Victoria NC, Inoue I, Young LJ, Murphy AZ. (2013a). A single neonatal injury induces life-long deficits in responses to stress. Dev Neurosci 35:326–37
- Victoria NC, Inoue K, Young LJ, Murphy AZ. (2013b). Long-term dysregulation of brain corticotrophin and glucocorticoid receptors and stress reactivity by single early-life pain experience in male and female rats. Psychoneuroendocrinology 38:3015–28
- Victoria NC, Inoue K, Young LJ, Murphy AZ. (2013c). A single neonatal injury induces life-long deficits in response to stress. Dev Neurosci 35:326–37
- Victoria NC, Karom MC, Eichenbaum H, Murphy AZ. (2014). Neonatal injury rapidly alters markers of pain and stress in rat pups. Dev Neurobiol 74:42–51
- Walker SM, Meredith-Middleton J, Cooke-Yarborough C, Fitzgerald M. (2003). Neonatal inflammation and primary afferent terminal plasticity in the rat dorsal horn. Pain 105:185–95
- Walker CD, Perrin M, Vale W, Rivier C. (1986a). Ontogeny of the stress response in the rat: role of the pituitary and the hypothalamus. Endocrinology 118:1445–51
- Walker CD, Sapolsky RM, Meaney MJ, Vale WW, Rivier CL. (1986b). Increased pituitary sensitivity to glucocorticoid feedback during the stress nonresponsive period in the neonatal rat. Endocrinology 119:1816–21
- Walter-Nicolet E, Annequin D, Biran V, Mitanchez D, Tourniaire B. (2010). Pain management in newborns: from prevention to treatment. Pediatr Drugs 12:353–65
- Wang G, Ji Y, Lidow MS, Traub RJ. (2004). Neonatal hind paw injury alters processing of visceral and somatic nociceptive stimuli in the adult rat. J Pain 5:440–9
- Watkins LR, Milligan ED, Maier SF. (2001). Glial activation: a driving force for pathological pain. Trends Neurosci 24:450–5
- Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF. (2015). Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male sprague dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35:316–24
- Weidenfeld J, Yirmiya R. (1996). Effects of bacterial endotoxin on the glucocorticoid feedback regulation of adrenocortical response to stress. Neuroimmunomodulation 3:352–7
- Whirledge S, Cidlowski JA. (2013). Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology 154:499–510
- Whirledge S, Xu X, Cidlowski JA. (2013). Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol Reprod 89:66 (1--17)
- Whitfield MF, Grunau RE. (2000). Behavior, pain perception, and the extremely low-birth weight survivor. Clin Perinatol 27:363–79
- Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. (2012). Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37:1491–505
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, et al. (2011). beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci 31:6277–88
- Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, Godbout JP. (2014). Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry 75:970–81
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. (2013). Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33:13820–33
- Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M, Rein T, et al. (2009). Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol Psychiatry 66:708–11